Abstract
Background
Cushing’s disease (CD) is rare in pediatric patients. It is characterized by elevated plasma adrenocorticotropic hormone (ACTH) from pituitary adenomas, with damage to multiple systems and development. In recent years, genetic studies have shed light on the etiology and several mutations have been identified in patients with CD.
Case presentation
A girl presented at the age of 10 years and 9 months with facial plethora, hirsutism and acne. Her vision and eye movements were impaired. A quick weight gain and slow growth were also observed. Physical examination revealed central obesity, moon face, buffalo hump, supra-clavicular fat pads and bruising. Her plasma ACTH level ranged between 118 and 151 pg/ml, and sella enhanced MRI showed a giant pituitary tumor of 51.8 × 29.3 × 14.0 mm. Transsphenoidal pituitary debulk adenomectomy was performed and immunohistochemical staining confirmed an ACTH-secreting adenoma. Genetic analysis identified a novel germline GPR101 (p.G169R) and a somatic USP8 (p. S719del) mutation. They were hypothesized to impact tumor growth and function, respectively.
Conclusions
We reported a rare case of pediatric giant pituitary ACTH adenoma and pointed out that unusual concurrent mutations might contribute to its early onset and large volume.
Background
Cushing’s disease (CD) is caused by the overproduction of adrenocorticotropic hormone (ATCH) by pituitary adenomas (PAs). It is rare in children and accounts for approximately 75% of pediatric Cushing’s syndrome from 7 to 17 years of age [1]. Weight gain and facial changes are more common in children than in adults [2]. Growth retardation is also a characteristic of children with hypercortisolemia [3]. Genetic alterations such as somatic USP8, RASD1, TP53 mutations, and germline AIP, MEN1, and CABLES1 mutations have been identified in CD patients [4]. Here we report a case of pediatric invasive pituitary ACTH macroadenoma associated with a novel germline GPR101 (p. G169R) and a somatic USP8 (p. S719del) mutation.
Case presentation
The girl was born at full term with a length of 48 cm and a weight of 2900 g. Her neuromotor and cognitive development was comparable to those of children of the same age. At the age of 9 years and 4 months she developed plethora, hirsutism, facial acne, rapid weight gain, and increased abdominal circumference. Her skin darkened, and purple striae appeared on thighs and in the armpits. She became dull and less talkative, as indicated by her parents. At 10 years and 3 months, the patient complained of pain around the left orbit with an intensity of 4–5 points on a numerical rating scale (NRS). Five months later bilateral blepharoptosis appeared, with significantly impaired vision of the left eye. Soon both eyes failed to rotate in all directions.
On admission the patient was 10 years and 9 months, with a height of 144 cm (90–97th percentile) and a weight of 48 kg (25–50th percentile). Her weight gain was 20 kg, while the height increased by only 2–3 cm in 18 months. Her blood pressure was 115/76mmHg, and her heart rate was 80 bpm. Apart from the signs mentioned above, physical examination revealed central obesity (BMI 23.1 kg/m2), moon face, buffalo hump, supra-clavicular fat pads and bruising at the left fossa cubitalis. Her pupils were 7 mm in diameter and barely reacted to light. There was a fan-shaped visual field defect in the left eye. Her breasts were Tanner stage III and pubic hair was Tanner stage II, although menarche had not yet occurred. The parents and her younger brother at 6 years of age did not have symptoms related to Cushing syndrome, acromegaly or gigantism. There was no family history of pituitary tumor or other endocrine tumors.
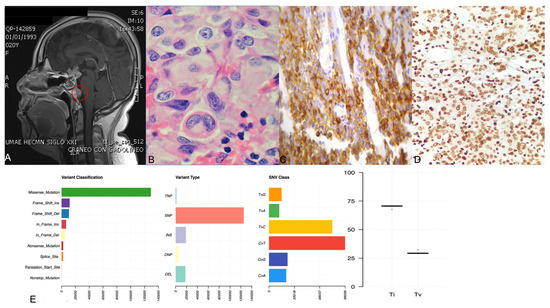
She had increased midnight serum cortisol (24.35 µg/dL, normal range < 1.8 µg/mL) and 24-hour urine free cortisol (24hUFC) (308.0 µg, normal range 12.3–103.5). The plasma ACTH level ranged from 118 to 151 pg/mL (< 46pg/mL). The 24hUFC was not suppressed (79.2 µg) after 48 h low-dose dexamethasone suppression test (LDDST), but suppressed to 32.8 µg (suppression rate 89.4%) after 48 h high-dose dexamethasone. Sella enhanced MRI showed a giant pituitary tumor measured 51.8 × 29.3 × 14.0 mm with heterogeneous density (Fig. 1). The mass compressed the optic chiasma and surrounded the bilateral cavernous sinus (Knosp 4). Therefore, an invasive giant pituitary ACTH adenoma was clinically diagnosed. The morning growth hormone (GH) was 1.0ng/ml (< 2 ng/ml) and insulin-like growth factor 1 416 ng/ml (88–452 ng/ml). The prolactin (PRL), luteinizing hormone (LH), follicle-stimulating hormone (FSH) and thyroid stimulating hormone (TSH) were all in normal ranges, as well as serum sodium, potassium, blood glucose and urine osmolality. Abdominal ultrasonography revealed a fatty liver. Tests concerning type 1 multiple endocrine neoplasia included serum calcium, phosphate, parathyroid hormone, gastrin and glucagon, which were all unremarkable (Table 1).
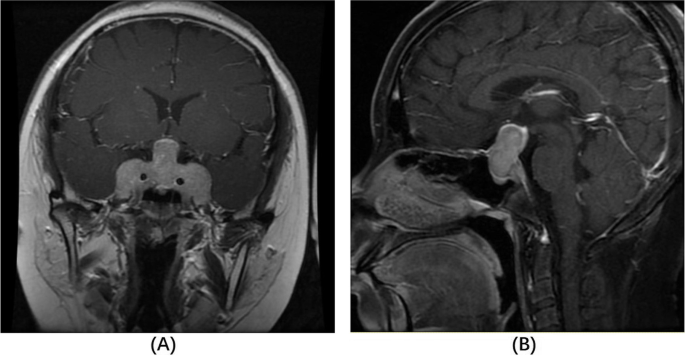
Contrast-enhanced coronal (A) and sagittal (B) T1-weighted MRI on admission. The sellar mass measured 51.8 × 29.3 × 14.0 cm (TD × VD × APD) with a heterogeneous density in the enhanced scan. The diaphragma sellea was dramatically elevated, with optic chiasm compressed. The sellar floor was sunken and bilateral cavernous sinus was surrounded (Knosp 4)
Transsphenoidal pituitary debulk adenomectomy was performed immediately due to multiple cranial nerve involvement and the negative results of Sandostatin loading test. A decompression resection was done. The plasma ACTH level declined to 77 pg/ml and serum cortisol 30.2 µg/dl three days after the operation. Vision, pupil dilation, eye movements and blepharoptosis also partially improved. Histopathology and immunohistochemical staining confirmed a densely–granulated corticotroph adenoma (Fig. 2, NanoZoomer S360 digital slide scanner and NDP.view 2.9.25 software, Hamamatsu, Japan). Neither necrosis nor mitotic activity was observed. The immunostaining for somatostatin receptor SSTR2A was positive with a cytoplasmic pattern, while GH, PRL, TSH, FSH, LH and PIT were all negative. The Ki 67 index was found to be 10%. One month after the operation the ACTH level increased to 132 pg/mL again, and the parents agreed to refer their child for radiotherapy to control the residual tumor.
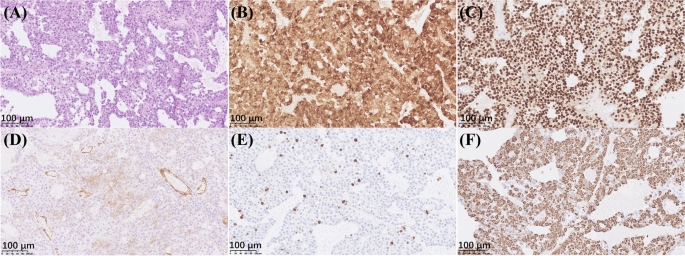
Histopathology and immunohistochemistry staining results of the pituitary tumor. By light microscopy, the tumor cells were mostly basophilic and arranged in papillary architecture. Neither necrosis nor mitotic activity was observed (A hematoxylin-eosin, ×200). Immunohistochemistry staining was positive for ACTH (B immunoperoxidase, ×200) and transcription factor T-PIT (C immunoperoxidase, ×200). Cytoplasmic staining of SSTR2A was observed in around 1/3 tumor cells besides the strong staining of endothelial cells (D immunoperoxidase, ×200). The Ki-67 index was 10% (E immunoperoxidase, ×200). Cytokeratin CAM5.2 was diffusely positive in the cytoplasm (F immunoperoxidase, ×200). The positive control for ACTH and T-PIT was the human anterior pituitary gland, and for SSRT2, Ki-67 and CAM5.2 were cerebral cortex, tonsil and colonic mucosa, respectively
The early onset and invasive behavior of this tumor led to the consideration of whether there was a genetic defect. Genetic studies were recommended for the families and they all agreed and signed the written informed consent forms. Whole exome sequencing (WES) was performed on the patient’s blood sample using an Illumina HiSeq sequencer to an average read depth of at least 90 times per individual. Raw sequence files were mapped to the GRCH37 human reference genome and analyzed using the Sentieon software. The results revealed a germline heterozygous GPR101 gene mutation c.505G > C (p.Gly169Arg), which was subsequently confirmed to be of maternal origin by Sanger sequencing. Meanwhile WES of the tumor tissue identified an additional somatic heterozygous c.2155_2157delTCC (p.S719del) mutation of the USP8 gene .
Discussion and conclusions
In this report, we described an extremely giant and invasive pituitary ACTH adenoma in a 10-year-old girl. According to Trouillas et al., invasive and proliferative pituitary tumors have a poor prognosis [5]. CD is rare among children, and the fast-growing and invasive nature of the tumor in this case led to the investigation of genetic causes. The somatic USP8 gene mutation has been recently reported to be associated with the pathogenesis of CD [6, 7]. This gene encodes ubiquitin-specific protease 8 (USP8). S718, S719 and P720 are hotspots in different studies [6,7,8,9,10,11,12,13,14]. They are located at the 14-3-3 binding motif, and the mutations disrupt the binding between USP8 and 14-3-3 protein, which leads to increased deubiquitination and EGFR signaling. High levels of EGFR consequently trigger proopiomelanocortin (POMC) transcription and ACTH secretion [6, 7]. The p.S719del mutation has been previously reported and its pathogenicity has been confirmed [7]. Thus, we speculate the p.S719del mutation plays a role in this patient with CD.
It is noteworthy that in our case, the pituitary corticotrophin adenoma was extremely giant and bilaterally invasive. USP8 mutations have been found in 31% of pediatric CD patients [10]. It is well known that microadenomas are most common in adult and pediatric CD patients. Previously, the Chinese and Japanese cohorts observed smaller sizes of USP8-mutated PAs than wild-type PAs [7, 9]. The Chinese cohort also reported a lower rate of invasive adenomas in USP8-mutated PAs [7]. This may be explained by the finding that UPS8 mutations did not significantly promote cell proliferation more than the wild-type ones [6]. Other cohorts suggested no difference in tumor size or invasiveness between USP8-mutated and wild-type PAs [8, 10, 12,13,14], which may be partially explained by the differences in sample sizes and ethnic backgrounds. Owing to the lack of evidence of USP8 mutations significantly contributing to tumor growth and invasiveness, additional pathogenesis should be investigated in this case.
The p.Gly169Arg mutation of the GPR101 gene has not been reported in patients with pituitary tumors. In silico predictions were performed using Polyphen-2, Mutation Taster and PROVEAN, and all of the programs reported it to be pathogenic. The GPR101 gene encodes an orphan G protein-coupled receptor (GPCR) and microduplication encompassing the gene has been proven to be the cause of X-linked acrogigantism (XLAG) [15]. XLAG is characterized by the early onset of pituitary GH-secreting macroadenomas. Point mutations of GPR101 have been found in patients with PAs that are mostly GH-secreting [15,16,17]. Although their prevalence is very low, an in vitro study supported the pathogenic role of p.E308D, the most common mutation of GPR101. This led to increased cell proliferation and GH production in rat pituitary GH3 cells [15]. Rare cases of PRL, ACTH or TSH-secreting PAs with GPR101 variants were also documented [16, 18]. To date, there have been five cases of ACTH-secreting PAs with four different germline GPR101 mutations: two cases of p.E308D, p.I122T, p.T293I and p.G31S, although in silico predictions and in vitro evaluations using AtT-20 cells have respectively determined the latter two mutations to be non-pathogenic [16, 18]. These patients were mainly children and young adults. Unlike pituitary GH-secreting tumors, the role of GPR101 mutations in the pathophysiology of CD is still questionable. Trivellin et al. demonstrated no statistically significant difference in GPR101 expression between corticotropinomas and normal human pituitaries. No significant correlation between GPR101 and POMC expression levels was found neither [18].
Given the evidences above, we hypothesize that the somatic USP8 mutation is responsible for the overexpression of ACTH in this CD girl while the germline GPR101 mutation contributes to the early onset and fast-growing nature of the tumor. Similarly, a 27-year-old woman with Nelson’s syndrome originally considered to be associated with a germline AIP variant (p.Arg304Gln) was recently reported to have a somatic USP8 mutation. The patient progressed rapidly and underwent multiple transsphenoidal surgeries [19]. Since germline AIP mutations are more commonly seen in GH-secreting PAs [20], the authors proposed that the USP8 mutation might have shifted the tumor towards ACTH-secreting [19]. Further investigations into the pathogenicity of GPR101 p.Gly169Arg and AIP p.Arg304Gln mutations are required to support the hypothesis.
In summary, we report a novel germline GPR101 and somatic USP8 mutation in a girl with an extremely giant pituitary ACTH adenoma. The concurrent mutations may lead to the growth and function of the tumor, respectively. Further investigations should be carried out to verify the role of the concurrent mutations in the pathogenesis of pediatric CD.
Availability of data and materials
The WES data of the blood sample of the patient is available in the NGDC repository (https://ngdc.cncb.ac.cn/gsa-human/) and the accession number is HRA002396. Any additional information is available from the authors upon reasonable request.
Abbreviations
- CD:
- Cushing’s disease
- ACTH:
- adrenocorticotropic hormone
- PA:
- pituitary adenoma
- NRS:
- numerical rating scale
- 24hUFC:
- 24-hour urine free cortisol
- LDDST:
- low-dose dexamethasone suppression test
- USP8:
- ubiquitin-specific protease 8
- POMC:
- proopiomelanocortin
- GPCR:
- G protein-coupled receptor
- XLAG:
- X-linked acrogigantism
References
-
Weber A, Trainer PJ, Grossman AB, Afshar F, Medbak S, Perry LA, et al. Investigation, management and therapeutic outcome in 12 cases of childhood and adolescent Cushing’s syndrome. Clin Endocrinol (Oxf). 1995;43(1):19–28.
-
Storr HL, Alexandraki KI, Martin L, Isidori AM, Kaltsas GA, Monson JP, et al. Comparisons in the epidemiology, diagnostic features and cure rate by transsphenoidal surgery between paediatric and adult-onset Cushing’s disease. Eur J Endocrinol. 2011;164(5):667–74.
-
Magiakou MA, Mastorakos G, Oldfield EH, Gomez MT, Doppman JL, Cutler GB Jr, et al. Cushing’s syndrome in children and adolescents. Presentation, diagnosis, and therapy. N Engl J Med. 1994;331(10):629–36.
-
Hernández-Ramírez LC, Stratakis CA. Genetics of Cushing’s Syndrome. Endocrinol Metab Clin North Am. 2018;47(2):275–97.
-
Trouillas J, Roy P, Sturm N, Dantony E, Cortet-Rudelli C, Viennet G, et al. A new prognostic clinicopathological classification of pituitary adenomas: a multicentric case-control study of 410 patients with 8 years post-operative follow-up. Acta Neuropathol. 2013;126(1):123–35.
-
Reincke M, Sbiera S, Hayakawa A, Theodoropoulou M, Osswald A, Beuschlein F, et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat Genet. 2015;47(1):31–8.
-
Ma Z-Y, Song Z-J, Chen J-H, Wang Y-F, Li S-Q, Zhou L-F, et al. Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res. 2015;25(3):306–17.
-
Perez-Rivas LG, Theodoropoulou M, Ferrau F, Nusser C, Kawaguchi K, Stratakis CA, et al. The Gene of the Ubiquitin-Specific Protease 8 Is Frequently Mutated in Adenomas Causing Cushing’s Disease. J Clin Endocrinol Metab. 2015;100(7):E997–1004.
-
Hayashi K, Inoshita N, Kawaguchi K, Ibrahim Ardisasmita A, Suzuki H, Fukuhara N, et al. The USP8 mutational status may predict drug susceptibility in corticotroph adenomas of Cushing’s disease. Eur J Endocrinol. 2016;174(2):213–26.
-
Faucz FR, Tirosh A, Tatsi C, Berthon A, Hernandez-Ramirez LC, Settas N, et al. Somatic USP8 Gene Mutations Are a Common Cause of Pediatric Cushing Disease. J Clin Endocrinol Metab. 2017;102(8):2836–43.
-
Albani A, Perez-Rivas LG, Dimopoulou C, Zopp S, Colon-Bolea P, Roeber S, et al. The USP8 mutational status may predict long-term remission in patients with Cushing’s disease. Clin Endocrinol (Oxf). 2018;89:454–8.
-
Ballmann C, Thiel A, Korah HE, Reis AC, Saeger W, Stepanow S, et al. USP8 Mutations in Pituitary Cushing Adenomas-Targeted Analysis by Next-Generation Sequencing. J Endocr Soc. 2018;2(3):266–78.
-
Losa M, Mortini P, Pagnano A, Detomas M, Cassarino MF, Pecori Giraldi F. Clinical characteristics and surgical outcome in USP8-mutated human adrenocorticotropic hormone-secreting pituitary adenomas. Endocrine. 2019;63(2):240–6.
-
Weigand I, Knobloch L, Flitsch J, Saeger W, Monoranu CM, Hofner K, et al. Impact of USP8 Gene Mutations on Protein Deregulation in Cushing Disease. J Clin Endocrinol Metab. 2019;104(7):2535–46.
-
Trivellin G, Daly AF, Faucz FR, Yuan B, Rostomyan L, Larco DO, et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med. 2014;371(25):2363–74.
-
Lecoq AL, Bouligand J, Hage M, Cazabat L, Salenave S, Linglart A, et al. Very low frequency of germline GPR101 genetic variation and no biallelic defects with AIP in a large cohort of patients with sporadic pituitary adenomas. Eur J Endocrinol. 2016;174(4):523–30.
-
Iacovazzo D, Caswell R, Bunce B, Jose S, Yuan B, Hernández-Ramírez LC, et al. Germline or somatic GPR101 duplication leads to X-linked acrogigantism: a clinico-pathological and genetic study. Acta Neuropathol Commun. 2016;4(1):56.
-
Trivellin G, Correa RR, Batsis M, Faucz FR, Chittiboina P, Bjelobaba I, et al. Screening for GPR101 defects in pediatric pituitary corticotropinomas. Endocr Relat Cancer. 2016;23(5):357–65.
-
Perez-Rivas LG, Theodoropoulou M, Puar TH, Fazel J, Stieg MR, Ferrau F, et al. Somatic USP8 mutations are frequent events in corticotroph tumor progression causing Nelson’s tumor. Eur J Endocrinol. 2018;178(1):57–63.
-
Tatsi C, Stratakis CA. The Genetics of Pituitary Adenomas. J Clin Med. 2019;9(1).
Acknowledgements
We thanked Dr. Xiaohua Shi and Dr. Yu Xiao from the Department of Pathology, Peking Union Medical College Hospital for their expertise in pituitary pathology and critical help in accomplishment of our manuscript.
Funding
This research was supported by “The National Key Research and Development Program of China” (No. 2016YFC0901501), “CAMS Innovation Fund for Medical Science” (CAMS-2017-I2M–1–011). They mainly covered the fees for genetic analysis and publications.
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Peking Union Medical College Hospital. The parents of the patient provided written informed consent for research participation.
Consent for publication
The parents of the patient provided written informed consent for the publication of indirectly identifiable data in this research.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
From https://bmcendocrdisord.biomedcentral.com/articles/10.1186/s12902-022-01058-8
Filed under: Cushing's, pituitary, Rare Diseases, symptoms | Tagged: ACTH, pediatric, pituitary | Leave a comment »