Early diagnosis and immediate treatment of Cushing’s syndrome (CS) are critical for a better prognosis but remain a challenge. However, few comprehensive reports have focused on this issue or investigated whether patient-reported manifestations are consistent with physician-assessed symptoms of CS. This study aimed to clarify the differences in patient-reported and physician-assessed manifestations of signs and symptoms of CS that prevent early diagnosis.
Methods
This single-center retrospective study included 52 patients with CS (16 with Cushing’s disease and 36 with adrenal CS). Upon clinical diagnosis, medical records were used to independently review the patient-reported and physician-assessed manifestations of typical (such as purple striae and proximal myopathy) and nonspecific features (such as hirsutism and hypertension). The correlations and differences between the patient-reported and physician-assessed manifestations were then analyzed.
Results
We observed a positive correlation between the total number of manifestations of nonspecific features reported by patients and those assessed by physicians, but not for typical features. Moreover, manifestations reported by the patients were less frequent than those assessed by physicians for typical features, leading to discrepancies between the two groups. In contrast, there were no differences in most nonspecific features between the patient-reported and physician-assessed manifestations. Notably, the concordance between patient-reported and physician-assessed manifestations of typical features was not associated with urinary free cortisol levels.
Conclusion
Regardless of disease severity, patients often do not complain of the typical features of CS that are crucial for formulating a diagnosis.
Introduction
Endogenous Cushing’s syndrome (CS) is caused by chronic and excessive glucocorticoid exposure. This occurs primarily due to adrenocorticotropic hormone (ACTH)-producing pituitary tumors (Cushing’s disease; CD) or cortisol-producing adrenal tumors (adrenal Cushing’s syndrome; ACS) [1]—and has a high mortality rate owing to cardiovascular disease, severe infection, and suicide, even when diagnosed and treated appropriately [1, 2]. Moreover, the prognosis is poor if the disease is not adequately treated or remains undiagnosed [2]. Therefore, early diagnosis and immediate intervention are important, as remission of CS due to surgical and pharmacological treatment can reduce the risk of mortality [3, 4].
CS is a rare disease with a prevalence of 57 per million individuals and an annual incidence of 3.2 per million, and its epidemiology is consistent across various regions worldwide [5, 6]. Most symptoms and signs of CS are common in general metabolic disorders, including obesity, hypertension, osteoporosis, and diabetes mellitus [7]. However, CS should be suspected if these symptoms appear as unusual features for their age [1, 8]. Consequently, the identification of CS is challenging and labor-intensive [1, 9, 10]. In fact, recent research revealed that a definitive diagnosis of CD (the most common form of CS), took an average of 3.8 ± 4.8 years from the onset of symptoms, and patients typically consulted 4.6 ± 3.8 medical professionals before this disease was identified [11]. Typical features of CS include symptoms of moon face, central obesity, or buffalo hump [12], which are similar to other symptoms such as primary obesity and therefore can lead to misdiagnosis. Furthermore, although purple striae or thin skin with an increased propensity for bruising are other typical features of CS [12], these attributes are not commonly acknowledged by the general population [1, 9].
Anzeige
Attempts have been made to diagnose CS early, including the development of scoring systems to estimate the pre-test probability of CS and facial image analysis software to diagnose the specific facial features of CS [13‐15]; however, these have not yet been used widespread or fully and the early diagnosis of CS remains dependent on the experience-based medical skills of the clinical staffs [16].
Additionally, although it is difficult for patients to recognize complex and nonspecific symptoms [17, 18], the significance of patients recognizing their illness has recently been reported for various diseases such as heart failure and malignant carcinoma [19‐21]. It is widely acknowledged that patients’ self-recognition can result in early detection of the disease, reduce its severity and recurrence, and enhance their quality of life [19]. In patients with endocrine diseases, there is increasing focus on issues surrounding self-recognition [22‐24]. For example, a previous study focusing on acromegaly reported a discrepancy between patient-reported and physician-reported manifestations and indicated that resolving this discrepancy could shorten the time to diagnosis [25].
Identifying CS may be challenging for primary care physicians who are yet to specialize. Therefore, endocrinologists with extensive experience in CS have often noticed that patients and these physicians struggle to identify the symptoms of CS; however, few comprehensive reports have focused on this issue or investigated whether patient-reported manifestations are consistent with physician-assessed symptoms of CS.
Therefore, this study aimed to investigate the unreported manifestations of CS among individuals referred to non-specialist healthcare providers, including primary care physicians, and to recognize potential challenges with the current diagnosis of CS with the goal of facilitating early detection.
Anzeige
Materials and methods
Patients, study design, and data collection
This single-center retrospective study was conducted to identify the discrepancies between patient-reported and physician-assessed symptoms and investigate the factors causing these differences.
From September 2004 to December 2022, 199 patients were referred to our department at a tertiary medical institution upon suspicion, evaluation, or follow-up for hypercortisolism. Of these patients, 92 were newly diagnosed with CS (36 with CD, 51 with ACS, and 5 with ectopic ACTH syndrome) based on the diagnostic guidelines [3, 8, 12], with a diagnosis confirmed by pathological evaluation after surgical resection [26]. However, 35 patients were excluded due to a lack of detailed clinical data on the manifestations at diagnosis. Similarly, we excluded individuals diagnosed with ectopic ACTH syndrome because of the lack of comprehensive information on symptoms reported by the patients and primary care physicians due to the rapid progression and severity of this disease. Therefore, 52 patients (16 with CD and 36 with ACS) were enrolled in this study.
Upon clinical diagnosis, the manifestations included in the comprehensive standardized interview at the time of diagnosis and those assessed by the physician through collaborative assessment with multiple board-certified endocrinologists as routine practice were independently reviewed from the medical records. We categorized these manifestations reviewed from the medical records into the following two categories based on the diagnostic guidelines including those of the Japan Endocrine Society: typical features, including moon face, central obesity or buffalo hump, purple striae of ≥1 cm, thin skin and easy bruising, and proximal myopathy; and nonspecific features (shown as atypical in Japan Endocrine Society’s guideline), including hypertension, menstrual abnormalities, acne, hirsutism, peripheral edema, glucose metabolism impairment, osteoporosis, pigmentation (which is not expected in patients with ACS), and mental abnormalities [1, 8, 12]. Central obesity or buffalo hump can also be observed in pseudo CS. However, in this study, features were classified as the same typical feature according to clinical guidelines [12, 27]. We also reviewed the biochemical findings, comorbidities, duration from the initial recognition of CS-related symptoms to diagnosis, and number of medical institutions visited before diagnosis.
The present retrospective study was performed in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Kobe University Hospital (Approval No. 1351). The patients had the option of an opt-out process, and all procedures were part of routine medical care.
Definition of patient-reported and physician-assessed manifestations
In the context of routine clinical care, physicians asked the patients about the presence or absence of manifestations and comorbidities (e.g., hypertension, menstrual abnormalities, glucose metabolism impairment, osteoporosis, and mental abnormalities), which were documented in the medical records. These reports in the medical records were defined as patient-reported manifestations in this study. In contrast, the manifestations and comorbidities of CS were assessed within several weeks after the patient was referred to our department for suspected CS. Additional diagnostic information on comorbidities is provided in the subsequent section. Physician-assessed manifestations were subsequently defined based on these findings.
Comorbidities of Cushing’s syndrome
All comorbidities were diagnosed according to the appropriate guidelines [28‐30]. For example, hypertension was diagnosed if patients were taking oral antihypertensive medication or had more than grade 1 hypertension (≥140/90 mmHg) in a treatment-naïve state [28]. Moreover, glucose metabolism impairment—including diabetes mellitus, impaired glucose tolerance, and impaired fasting glucose—was diagnosed based on the results of blood glucose levels during fasting and after a 75-g oral glucose tolerance test, as well as hemoglobin A1c (HbA1c) levels [29]. Patients taking medications for diabetes mellitus at the time of CS diagnosis were also categorized as having diabetes.
Other comorbidities included mental abnormalities, menstrual abnormalities, and the presence of osteoporosis. Mental abnormalities were defined as the use of anxiolytic medications, sleeping pills, or antidepressants prescribed by experienced psychologists, and menstrual abnormalities were defined as women with irregular menstrual cycles. Furthermore, the presence of osteoporosis was defined as bone mineral density (BMD) of <–2.5 standard deviations (SD) of the T-score of the lumbar vertebrae (L2–L4), femoral neck, or distal radius measured using dual-energy x-ray absorptiometry (DXA; Horizon A DXA System), and/or an experience of a fragility fracture [30]. As per the specifications of the measurement system employed, L1 was not included in the assessment. The Z-score was also employed as a diagnostic reference among young adults. Patients also diagnosed with osteoporosis who were receiving medications for this disease.
Hormone assay
In this study, blood samples were collected after an overnight fast. Subsequently, serum cortisol levels were measured using a chemiluminescent enzyme immunoassay [CLEIA] (TOSOH, Tokyo, Japan, RRID:AB_3099658) or enzyme immunoassay [EIA] (TOSOH, Tokyo, Japan, RRID:AB_3076600). Similarly, plasma ACTH levels were measured using a CLEIA (TOSOH, Tokyo, Japan, RRID:AB_3099657, or Siemens, Tokyo, Japan, RRID:AB_2909441) and EIA (TOSOH, Tokyo, Japan, RRID:AB_2783633). In both methods, the measurements showed good correlation and no conversion was required [31, 32].
Anzeige
Urinary free cortisol (UFC) levels were also measured using radioimmunoassays (RIA; TFB, Tokyo, Japan, RRID:AB_2894408) or chemiluminescent immunoassays (CLIA; Siemens, Tokyo, Japan, RRID:AB_2893154). Using the following formula, the UFC levels measured by RIA were then corrected to the value measured by CLIA: Y = 0.832X − 4.23 (Y = UFC levels using CLIA, X = UFC levels using RIA) [33].
Statistical analysis
All statistical analyses were performed using SPSS ver. 28.0 software (IBM Corp., Armonk, NY, USA). All continuous variables were analyzed using the Shapiro–Wilk normality test to confirm a normal distribution, whereas Fisher’s exact test was used to analyze categorical data. Between the two groups, differences in normally or non-normally distributed data were compared using the unpaired Student’s t-test or the Mann–Whitney U test, respectively.
Cohen’s kappa coefficient was used to describe the concordance between the patient-reported and physician-assessed manifestations. As previously reported [19, 20, 34], the concordance based on the value of Cohen’s kappa coefficient was rated as follows: 0.00–0.20 for “Slight,” 0.21–0.40 for “Fair,” 0.41–0.60 for “Moderate,” 0.61–0.80 for “Substantial,” and 0.81–1.00 for “Almost Perfect.” For correlation analysis between two variables of non-normally distributed data, we used Spearman’s rank correlation coefficient. Multivariate logistic regression analyses were then performed to investigate variables associated with the discrepancies between patient-reported and physician-assessed manifestations.
The results are presented as mean ± SD for normally distributed data and median [interquartile range] for non-normally distributed data, and differences were considered statistically significant when the P value was <0.05.
Anzeige
Results
Clinical characteristics of the patients
We included 52 patients diagnosed with CS in this study. Their clinical characteristics are presented in Table 1. Notably, this group consisted of 5 males and 47 females, with a mean age of 49.4 ± 15.8 years, median body mass index (BMI) of 23.0 [21.3–28.0] kg/m2, and median UFC level of 272.1 [126.0–435.0] µg/day. Of the CS patients, 16 had CD and 36 had ACS, which is consistent with epidemiological data on CS observed in Asians (including Japanese individuals); however, this differed from epidemiological data from Western countries [35, 36]. Regarding comorbidities, 43 patients were diagnosed with hypertension—of which 34 were prescribed antihypertensive medications—with a mean systolic blood pressure (BP) of 136.4 ± 21.5 mmHg and diastolic BP of 83.5 ± 15.0 mmHg. In addition, 44 patients were diagnosed with glucose metabolism impairment—of which, 20 were prescribed oral hypoglycemic agents and/or insulin—with a median fasting serum glucose level of 99.5 [87.3–116.5] mg/dL and median HbA1c level of 6.3% [5.7–7.4]. Moreover, 29 patients were diagnosed with osteoporosis, of which 4 were prescribed antiosteoporosis medication, with BMD T-score SDs of -1.54 ± 1.39, -1.76 ± 1.12, and -0.50 [-1.53–0.50] for the lumber spine, femoral neck, and distal radius, respectively. Notably, the UFC levels were higher in patients with CD than in those with ACS (412.6 [243.2–1,100.3] vs. 215.3 [114.0–387.8] µg/day); however, there were no significant differences attributed to sex, age, BMI, or the proportion of patients with respect to comorbidities, including hypertension and glucose metabolism impairment, between patients with CD and ACS.
Table 1
Clinical characteristics of the patients
Total
CD
ACS
CD vs. ACS P value
Number of men/women
5/47
1/15
4/32
1.00
Age (years)
49.4 ± 15.8
54.3 ± 19.2
47.2 ± 13.8
0.14
BMI (kg/m2)
23.0 [21.3–28.0]
24.7 [22.2–30.0]
22.8 [20.8–26.4]
0.17
Midnight F (µg/dL)
20.1 [16.0–23.5]
20.2 [13.9–24.7]
20.1 [16.9–23.0]
0.97
F after LDDST (μg/dL)
21.2 ± 6.9
24.2 ± 10.1
19.7 ± 4.2
0.11
UFC (μg/day)
272.1 [126.0–435.0]
412.6 [243.2–1,100.3]
215.3 [114.0–387.8]
0.02
Basal ACTH (pg/mL)
2.0 [0.0–53.9]
83.2 [57.4–169.9]
0.0 [0.0–2.1]
<0.01
Systolic BP (mmHg)
136.4 ± 21.5
140.5 ± 20.7
134.6 ± 21.8
0.36
Diastolic BP (mmHg)
83.5 ± 15.0
83.1 ± 14.3
83.6 ± 15.5
0.90
Use of antihypertensive drugs, n (%)
34 (65)
13 (81)
21 (58)
0.13
FSG (mg/dL)
99.5 [87.3–116.5]
110.0 [102.0–142.8]
92.5 [83.3–114.3]
0.01
HbA1c (%)
6.3 [5.7–7.4]
6.8 [5.9–8.6]
6.0 [5.7–7.1]
0.08
Use of OHA and/or insulin, n (%)
20 (38)
9 (56)
11 (31)
0.12
LS BMD T-score (SD)
−1.54 ± 1.39
−1.00 ± 1.38
−1.79 ± 1.35
0.07
LS BMD Z-score (SD)
−0.78 ± 1.37
0.13 ± 1.11
−1.20 ± 1.28
<0.01
FN BMD T-score (SD)
−1.76 ± 1.12
−1.73 ± 1.54
−1.78 ± 0.88
0.92
FN BMD Z-score (SD)
−0.79 ± 1.01
−0.39 ± 1.10
−0.99 ± 0.92
0.10
Radius BMD T-score (SD)
−0.50 [−1.53–0.50]
−0.30 [−2.50–0.40]
−0.60 [−1.30–0.60]
0.79
Radius BMD Z-score (SD)
0.60 [−0.60–1.50]
1.50[−0.60–1.80]
0.50[−0.50–1.00]
0.33
Use of antiosteoporosis drugs, n (%)
4 (8)
1 (6)
3 (8)
1.00
Time to diagnosis (months)
44.0 [13.3–125.3]
43.0 [15.0–128.3]
47.5 [12.5–125.3]
0.87
Number of medical institutions before diagnosis
3.0 [2.0–5.0]
3.0 [2.0–5.0]
3.0 [3.0–5.8]
0.23
The results are presented as mean ± SD for normally distributed data and median [interquartile range] for non-normally distributed data
CD Cushing’s disease, ACS adrenal Cushing’s syndrome, BMI body mass index, F cortisol, LDDST low-dose dexamethasone suppression test, UFC urinary free cortisol, ACTH adrenocorticotropic hormone, BP blood pressure, FSG fasting serum glucose, HbA1c hemoglobin A1c, OHA oral hypoglycemic agents, BMD bone mineral density, LS lumber spine, FN femoral neck
The median duration from the patients’ initial recognition of CS-related manifestations to diagnosis was 44.0 [13.3–125.3] months, and it took more than 3 years to diagnose CS in 30 patients (58%). Furthermore, the median number of medical facilities visited by patients before diagnosis was 3.0 [2.0–5.0]; however, there were no significant differences in the duration or number of medical institutions between patients with CD and those with ACS.
Frequency and concordance between patient-reported and physician-assessed CS-related manifestations
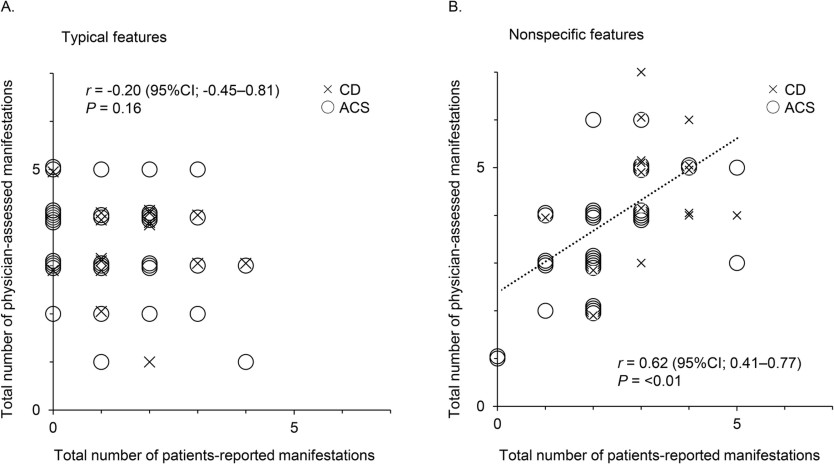
Each manifestation reported by a patient or assessed by a physician is shown vertically for individual cases in Fig. 1. Compared with nonspecific features, typical features appeared to not be reported by the patients but were only assessed by the physicians. In addition, compared to nonspecific features, there were fewer cases in which the manifestations reported by the patients were consistent with those assessed by physicians for typical features.
Fig. 1
Consistency between patient-reported and physician-assessed manifestations for each individual case. The consistencies or discrepancies between patient-reported and physician-assessed manifestations are shown. Vertical lines represent manifestations in individual patients. CD Cushing’s disease, ACS adrenal Cushing’s syndrome
Consistent with the impact of these visually distinctive presentations shown in Fig. 1, no correlation was observed in the number of typical features between patient-reported and physician-assessed manifestations (r = –0.20, P = 0.16) (Fig. 2A), whereas a positive correlation was found for nonspecific features (r = 0.62, P < 0.01) (Fig. 2B). Moreover, the total number of patient-reported manifestations of typical features was lower than that of physician-assessed manifestations (1.0 [0.0–2.0] vs. 3.5 [3.0–4.0], P < 0.01), and four of the five typical features were reported less frequently by patients than by physicians, except for proximal myopathy (Table 2A). According to Cohen’s kappa coefficient, the concordance between patient-reported and physician-assessed manifestations was marked as “Fair” to “Slight,” indicating a discrepancy for all typical features. Similarly, the total number of patient-reported manifestations of nonspecific features was also lower than that in physicians (2.5 [2.0–3.0] vs. 4.0 [3.0–5.0], P < 0.01). However, except for glucose metabolism impairment or osteoporosis, there were no differences in the frequencies of nonspecific features between patient-reported and physician-assessed manifestations, and the concordance of the nonspecific features between the patient-reported and physician-assessed manifestations was “Almost perfect” for menstrual abnormality and “Substantial” for mental abnormality and hypertension, whereas that for glucose metabolism impairment and osteoporosis was “Fair.” This suggests that the discrepancy between patient-reported and physician-assessed manifestations was more significant for typical than for nonspecific features. However, no differences in these discrepancies were observed between patients with CD and those with ACS (Table 2B, C).
Fig. 2
Correlation between the total number of patient-reported and physician-assessed manifestations. Correlations between the total number of patient-reported and physician-assessed manifestations are shown for typical (A) and nonspecific features (B). CD is plotted by ×, and ACS is plotted by ○. The Spearman’s rank correlation coefficients and P value are presented. CI confidence interval, CD Cushing’s disease, ACS adrenal Cushing’s syndrome
Table 2
Frequencies of patient-reported and physician-assessed manifestations and their concordance. A. All patients (n = 52). B. Patients with CD (n = 16). C. Patients with ACS (n = 36)
Patient-reported
Physician-assessed
P value of Fisher’s exact test
Concordance with Cohen’s kappa coefficient
A
Typical features
Moon face, n (%)
20 (39)
48 (92)
<0.01
Slight
Central obesity or buffalo hump, n (%)
13 (25)
44 (85)
<0.01
Slight
Purple striae, n (%)
3 (6)
15 (29)
<0.01
Fair
Thin skin and easy bruising, n (%)
15 (29)
43 (83)
<0.01
Slight
Proximal myopathy, n (%)
21 (40)
27 (52)
0.33
Slight
Nonspecific features
Hypertension, n (%)
39 (75)
43 (83)
0.47
Substantial
Menstrual abnormalities, n (%)
11 (21)
11 (21)
1.00
Almost perfect
Acne, n (%)
7 (14)
13 (25)
0.21
Moderate
Hirsutism, n (%)
3 (6)
10 (19)
0.07
Moderate
Peripheral edema, n (%)
24 (46)
28 (54)
0.56
Fair
Glucose metabolism impairment, n (%)
24 (46)
44 (85)
<0.01
Fair
Osteoporosis, n (%)
7 (14)
29 (56)
<0.01
Slight
Pigmentation, n (%)
0 (0)
5 (10)
0.06
–
Mental abnormalities, n (%)
17 (33)
17 (33)
1.00
Substantial
B
Typical features
Moon face, n (%)
6 (38)
14 (88)
0.01
Slight
Central obesity or buffalo hump, n (%)
6 (38)
15 (94)
<0.01
Slight
Purple striae, n (%)
2 (13)
4 (25)
0.56
Moderate
Thin skin and easy bruising, n (%)
4 (25)
13 (81)
0.06
Slight
Proximal myopathy, n (%)
8 (50)
8 (50)
1.00
Slight
Nonspecific features
Hypertension, n (%)
16 (100)
15 (94)
0.78
Slight
Menstrual abnormalities, n (%)
5 (31)
5 (31)
1.00
Almost perfect
Acne, n (%)
1 (6)
3 (19)
0.56
Moderate
Hirsutism, n (%)
2 (13)
4 (25)
0.56
Moderate
Peripheral edema, n (%)
8 (50)
10 (63)
0.56
Slight
Glucose metabolism impairment, n (%)
10 (63)
15 (94)
0.14
Slight
Osteoporosis, n (%)
4 (25)
9 (56)
0.15
Slight
Pigmentation, n (%)
0 (0)
5 (31)
0.14
–
Mental abnormalities, n (%)
5 (31)
6 (38)
0.78
Moderate
C
Typical features
Moon face, n (%)
14 (39)
34 (94)
<0.01
Slight
Central obesity or buffalo hump, n (%)
7 (19)
29 (81)
<0.01
Slight
Purple striae, n (%)
1 (3)
11 (31)
<0.01
Slight
Thin skin and easy bruising, n (%)
11 (31)
30 (83)
<0.01
Slight
Proximal myopathy, n (%)
13 (36)
19 (53)
0.24
Slight
Nonspecific features
Hypertension, n (%)
23 (64)
28 (78)
0.30
Substantial
Menstrual abnormalities, n (%)
6 (17)
6 (17)
1.00
Almost perfect
Acne, n (%)
6 (17)
10 (28)
0.40
Moderate
Hirsutism, n (%)
1 (3)
6 (17)
0.11
Fair
Peripheral edema, n (%)
16 (44)
18 (50)
0.81
Fair
Glucose metabolism impairment, n (%)
14 (39)
29 (81)
<0.01
Fair
Osteoporosis, n (%)
3 (8)
20 (56)
<0.01
Slight
Pigmentation, n (%)
0 (0)
0 (0)
–
–
Mental abnormalities, n (%)
12 (33)
11 (31)
1.00
Almost perfect
The frequencies of patient-reported and physician-assessed manifestations were compared using Fisher’s exact test. The concordance between patient-reported and physician-assessed manifestations was evaluated with Cohen’s kappa coefficient, and its coefficients were defined as follows: 0.00–0.20 for “Slight,” 0.21–0.40 for “Fair,” 0.41–0.60 for “Moderate,” 0.61–0.80 for “Substantial,” and 0.81–1.00 for “Almost perfect”
CD Cushing’s disease, ACS adrenal Cushing’s syndrome
Anzeige
We performed logistic regression analyses using UFC to investigate whether excess cortisol levels influenced the discrepancy between patient-reported and physician-assessed manifestations. Notably, we observed no association between UFC levels and discrepancies between patient-reported and physician-assessed manifestations in the univariate or multivariate logistic regression analyses adjusted for sex and age (Table 3A). In addition, no association was observed after adjusting for other variables such as BMI and disease duration. Similarly, we found that the serum cortisol levels after the low-dose dexamethasone suppression test (LDDST) were not associated with discrepancies between patient-reported and physician-assessed manifestations (Table 3B). Thus, these disparities were shown to be insignificant when directly related to the severity of CS.
Table 3
Logistic regression analyses of the discrepancies between the patient-reported and physician-assessed manifestations. A. Variables associated with UFC levels. B. Variables associated with serum cortisol levels after the LDDST
Univariate
Multivariate 1 (sex- and age-adjusted)
Multivariate 2 (BMI-adjusted)
Multivariate 3 (disease duration-adjusted)
A
Moon face
1.000 (0.999–1.001)
1.000 (0.999–1.001)
1.000 (0.998–1.002)
1.000 (0.999–1.001)
Proximal myopathy
1.000 (0.999–1.001)
1.000 (0.999–1.001)
1.000 (0.998–1.001)
1.000 (0.998–1.001)
Thin skin and easy bruising
1.000 (0.998–1.001)
1.000 (0.999–1.001)
1.000 (0.999–1.001)
1.000 (0.998–1.001)
Central obesity or buffalo hump
1.001 (1.000–1.003)
1.001 (1.000–1.003)
1.001 (1.000–1.003)
1.001 (1.000–1.003)
Purple striae
1.000 (0.999–1.002)
1.000 (0.998–1.002)
1.001 (0.999–1.003)
1.000 (0.999–1.002)
B
Moon face
0.998 (0.919–1.084)
0.999 (0.919–1.086)
1.000 (0.920–1.088)
0.997 (0.918–1.082)
Proximal myopathy
1.007 (0.925–1.097)
1.007 (0.924–1.097)
1.007 (0.925–1.097)
1.006 (0.924–1.096)
Thin skin and easy bruising
1.022 (0.939–1.112)
1.018 (0.934–1.109)
1.023 (0.940–1.113)
1.019 (0.937–1.109)
Central obesity or buffalo hump
0.979 (0.890–1.078)
0.978 (0.865–1.105)
0.981 (0.875–1.099)
0.978 (0.887–1.078)
Purple striae
0.998 (0.919–1.084)
0.999 (0.919–1.086)
1.000 (0.920–1.088)
0.997 (0.918–1.082)
The results are presented as odds ratios (95% confidence intervals)
UFC urinary free cortisol, BMI Body Mass Index, LDDST low-dose dexamethasone suppression test
Discussion
In the present study, we highlight the challenges associated with the diagnosis of CS—a condition resulting from excessive glucocorticoid exposure—and elucidate the divergence between patient-reported and physician-assessed manifestations. Thus, this study may aid in the early detection of CS by identifying symptoms that patients are unable to recognize based on the disparities between patient-reported and physician-assessed manifestations of CS.
In this study, the number of patient-reported manifestations of both typical and nonspecific features was lower than that of physician-assessed manifestations, suggesting that CS symptoms may have been overlooked by relying solely on patient reports. Additionally, analysis of the concordance between patient-reported and physician-assessed manifestations revealed a tendency for these manifestations to be inconsistent for both typical and nonspecific features, with a tendency to be more significant for typical features. Furthermore, the UFC and serum cortisol levels after the LDDST, which represent the severity of CS, were not associated with the concordance of manifestations between patients and physicians, suggesting that even in cases of severe CS, patients may not recognize their symptoms. These findings imply that typical features, which are essential for diagnosing CS, may be difficult for patients to recognize and poorly identified or conveyed to patients by non-specialist physicians, who are typically the first to interact with individuals with CS. The importance of educating healthcare providers such as primary care physicians, family physicians and gynecologists for early diagnosis of CS should be highlighted.
According to a previous report on the diagnostic history of 176 patients with CD, 83% of the patients visited their family physician for manifestations such as weight gain and hypertension, while 46% visited a gynecologist for menstrual abnormalities before the diagnosis of CD [11]. Thus, the typical features of CS were not recognized. The examination may reveal nonspecific features. However, individuals who are non-specialists may not recognize these features as indications of CS. Therefore, patients are often unaware of the potential complications associated with CS. This is consistent with the results of our study, in which patient-reported and physician-assessed manifestations were more consistent for hypertension and menstrual abnormalities than for other manifestations such as typical features, glucose metabolism impairment, and osteoporosis. This makes diagnosis challenging as non-specialist physicians and, more prominently, patients may not recognize the full range of symptoms associated with CS, especially the typical features with high diagnostic value. In addition, older patients diagnosed with CS present with a lower BMI and waist circumference than younger patients [37], and they typically do not exhibit symptoms commonly associated with CS such as skin alterations, depression, hair loss, hirsutism, and reduced libido. These findings may further complicate the diagnosis of CS in elderly patients.
By evaluating only the patient-reported manifestations, it appears that manifestations such as peripheral edema and proximal myopathy were more common. Possibly, these symptoms were not considered features of CS by physicians, in comparison to the degree of symptoms experienced by the patients. However, this may not necessarily imply diminishing the significance of the patient’s signs and symptoms, as these manifestations can be considered as the unidentified complaints and may result in a postponement of the diagnosis of CS. Patients may be experiencing symptoms that physicians do not perceive, indicating the importance of interview and physical examination. Further investigation is needed to elucidate underlying factors.
Considering the rarity of CS, it is crucial to suspect and diagnose the condition based on clinical symptoms and perform the appropriate screening tests without over- or under-screening [7]. Although CS screening in patients with diabetes mellitus and hypertension has been reported to lead to a diagnosis in only 0–0.7% and 0.1–0.5% of these patients, respectively [38‐41], it is ineffective in terms of false positives and cost [9]. Therefore, patients with typical features that are highly specific for CS, such as purple striae, easy bruising, and proximal myopathy [1, 8, 12], as well as those with obesity, diabetes mellitus, or hypertension in combination with these features, should be screened for CS [7, 27]. However, our results suggest that these symptoms are unlikely to be self-recognized. Therefore, the appropriate screening measures must be implemented to establish an early and effective diagnosis of CS.
In these situations, it is crucial for physicians to utilize their knowledge and experience to suspect CS based on symptoms such as typical features [10]. It has been reported that years of clinical experience in endocrine practice can contribute to the estimation of the pre-test probability of CS [16]. In contrast, non-specialists are less likely to encounter patients with CS in their lifetime, which can make it difficult to properly suspect CS [9]. From this perspective, it is of utmost importance that family physicians and general internists are knowledgeable regarding the manifestations that require screening for CS, as early diagnosis of this uncommon and severe condition is crucial [11]. Therefore, it is important for physicians who routinely treat patients presenting with common symptoms such as obesity, diabetes mellitus, and hypertension to meticulously interview and observe for any indicators of CS, even if the patient does not recognize them. Failure to adopt an appropriate tone in these situations may cause the disease to become undetectable.
In rare disorders such as CS, in addition to enhancing public recognition of the disease, the appropriate sharing of information and provision of specialized care in clinical practice remain important issues [42]. Early identification of such rare diseases can be achieved by promoting an understanding of the disease and its symptoms among family, friends, and patients who may be the first to recognize the signs and symptoms in an individual. In fact, in a questionnaire survey of 340 patients with CS across 30 countries, the diagnosis of CS was made in 5.6% of cases by the patients themselves and in 0.9% by their family or friends [43]. In the present study, we found that it took more than 3 years to diagnose CS in 58% of the cases. If CS and its symptoms are popularized among the public, the typical features of CS could be more readily reported to physicians and the time to diagnosis might be shorter. Furthermore, a primary care physician who is well-educated and knowledgeable is crucial in ensuring that the concerns of such individuals are not overlooked.
This study has some limitations. First, this single-center retrospective study included a relatively small sample size with few male patients. Second, CD and ACS have different pathologies; therefore, the frequencies of several CS-related manifestations will differ depending on their subtypes [3, 44]. However, in this study, there was no difference in the discrepancies between patient-reported and physician-assessed manifestations in patients with CD or ACS. Nonetheless, it is crucial that comprehensive research is conducted in larger patient populations with a focus on employing methods that accurately reflect the pathophysiology of CD and ACS. Third, patient reports may be inaccurate in terms of onset and duration because they depend on the patient’s memory. Fourth, the endocrinologists who examined the patients differed, which may have affected the presence or absence of physician-assessed manifestations. Finally, this study investigated the differences between the manifestations reported by patients and those assessed by endocrinologists, although the evaluations conducted by primary care physicians, which are crucial for the early detection of CS, were not available. Future research is needed to investigate the differences in recognizing manifestations between non-specialist physicians and endocrinologists with extensive experience in CS and to examine the changes before and after education for these non-specialists to determine if they can lead to earlier diagnosis of CS.
In conclusion, endocrinologists have been shown to be aware of CS-related symptoms, especially typical features, whereas patients do not recognize these manifestations, even when the disease is severe. Therefore, the key to the early diagnosis and treatment of CS is a more proactive approach of questioning and examining patients suspected of having the disease.
Acknowledgements
We thank all the physicians and medical assistants who were involved in this study. We are grateful to all the laboratory members for their excellent discussions and fruitful suggestions. We also thank Editage (www.editage.jp) for English language editing.
Compliance with ethical standards
Conflict of interest
The authors declare no competing interests.
Ethics approval
This study conformed to the Declaration of Helsinki guidelines and was approved by the Ethics Committee of Kobe University Hospital (Approval No. 1351).
Informed consent
Informed consent was obtained from all the participants using an opt-out approach.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Activating mutation in Ubiquitin-specific peptidase (USP8) is identified to enhance cell proliferation and adrenocorticotropic hormone (ACTH) secretion from corticotroph pituitary adenoma. We investigated the USP8 variant status in a population of Iranian people with functional corticotroph pituitary adenoma (FCPA). Moreover, a systematic review was conducted to thoroughly explore the role of USP8 variants and the related pathways in corticotroph adenomas, genotype-phenotype correlation in USP8-mutated individuals with FCPA, and the potential role of USP8 and epidermal growth factor receptor (EGFR) as targeted therapies in PFCAs.
Methods
Genetic analysis of 20 tissue samples from 19 patients with PFCAs was performed using Sanger sequencing. Moreover, a systematic literature review was performed using the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines. PubMed, Scopus, web of Sciences, and Cochrane databases were searched. The last search was performed on 20 September 2023 for all databases.
Results
In our series, we found two somatic mutations including a 7-bp deletion variant: c.2151_2157delCTCCTCC, p. Ser718GlnfsTer3, and a missense variant: c.2159 C > G, p. Pro720Arg (rs672601311) in exon 14. The Systematic review indicated USP8 variant in 35% of corticotroph adenomas, with the highest frequency (25%) in 720 code regions, p. Pro720Arg. Data regarding the impact of USP8 mutational status on clinical characteristics and outcomes in FCPAs are inconsistent. Moreover, Pasireotide as well as inhibitors of EGFR such as Gefitinib and Lapatinib, as well as USP8 inhibitors including -ehtyloxyimino9H-indeno (1, 2-b) pyrazine-2, 3-dicarbonitrile, DUBs-IN-2, and RA-9 indicated promising results in treatment of corticotroph adenomas.
Conclusion
Although the USP8–EGFR system has been identified as the main trigger and target of corticotroph tumorigenesis, more precise multicenter studies are required to yield more consistent information regarding the phenotype-genotype correlation and to develop effective targeted therapies.
Pituitary corticotroph adenoma accounts for 68% of endogenous hypercortisolism [1]. Prolonged exposure to high cortisol levels is associated with a variety of long-term complications, impaired quality of life, and increased mortality [2]. Transsphenoidal surgical excision is the treatment of choice. However, curative surgery is challenging with the initial remission rate of 65–85% and a high recurrence rate [3, 4].
The majority of functional corticotroph adenomas (FCAs) are sporadic. Although the genetic background is not well-established, potential candidate genes are proposed for tumor initiation and progression [5]. Hotspot mutations in ubiquitin-specific peptidase (USP8) are reported in 11–62% of sporadic corticotroph adenomas [6,7,8]. USP8 is a deubiquitinating enzyme that plays an important role in enhancing cell proliferation and regulating cell cycle [9]. The mutant USP8 was found to activate the epidermal growth factor receptor (EGFR) signaling pathway ultimately promoting adrenocorticotrophic hormone (ACTH) secretion [6]. Moreover, overexpression of EGFR and its signaling pathway components in pituitary corticotroph adenoma was reported [10]; and found to be positively associated with ACTH and cortisol levels as well as tumor recurrence [10]. These outcomes suggest that USP8 and EGFR are promising biomarkers for prediction of recurrence and can be used as targeted therapy.
Thus, we conducted a study to examine the USP8 and ubiquitin-specific peptidase 48 (USP48) variations in a group of Iranian people with Cushing’s disease (CD) and carried out a systematic review of the literature regarding the USP8/EGFR and their potential role in the clinical outcomes and targeted therapy in CD.
Methods
Case series
Study population
Paraffin-embedded blocks of pituitary tumor tissue from 19 patients with ACTH-secreting pituitary adenoma who underwent transsphenoidal surgery (TSS) between 2011 and 2019 were examined. The diagnosis of CD was based on clinical features and biochemical criteria [11]. The patients clinically suspected to CD were asked to collect urine free cortisol (UFC) in two separated times and underwent overnight dexamethasone suppression test (ODST). After confirmation of ACTH-dependent Cushing’s syndrome using measurement of ACTH level, a high-dose dexamethasone suppression test (HDDST) was performed to confirm the pituitary source of hypercortisolism. Patients with equivocal results or those with pituitary tumors less than 6 mm in size were undergone inferior petrosal sinus sampling (IPSS). Patient with clinical, biochemical, and radiological evidences of CD were undergone TSS. And eventually, corticotroph adenoma was confirmed using immunohistochemically staining of tumor tissue in all patients. The study was approved by the IUMS Research Ethics Committee (IR.IUMS.REC.1398.082). It was carried out under the declaration of Helsinki and the International Conference on Harmonization of Good Clinical Practice (ICH-GCP) guidelines, and informed consent was obtained from all patients.
DNA extraction and Sanger sequencing
A 10-µm thick section of formalin-fixed and paraffin-embedded (FFPE) tissue per sample was used for genomic DNA extraction. A molecular test was performed by amplification of USP8 and USP48 hotspot exons (exon 14 and exon 10, respectively) using conventional polymerase chain reaction (PCR). USP8 was amplified by two primer pairs; USP8_F1: AGCAGAATACTTTGGAGTGATTTC and USP8_R1: TTTGGAAGGTTCCCTATCCC with 251 bp product, USP8_F2: ACCCCTCCAACTCATAAAGC and USP8_R2: GAGTAGAAACTTTGAAATACAGCAC, with 220 bp product. A 240 bp fragment of USP48 was produced using; USP48_F: CCCGCTAAAGAATAAACAAACTC and USP48_R: GCATTCTAAAACATTTGCCTGC. PCR was done in 25 µl final volume (Ampliqon 2x PCR Mix) containing 0.5 µM of each primer and 30 ng of genomic DNA for 35 cycles (94 °C for 20 s, annealing 60 °C for 20 s and extension 72 °C for 20 s). The quality of PCR products was assessed by 2% agarose gel electrophoresis. Bidirectional Sanger sequencing was performed on an ABI DNA Analyzer (Applied Biosystems), The PCR primers were also used in the sequencing reaction. CodonCode Aligner software was used to analyze hotspot exome sequencing. Sequencing data quality was evaluated using Sanger electropherograms of both forward and reverse strands. The identified somatic mutations were analyzed in DNA taken from whole blood samples, but germline mutation was not detected.
Systematic review
Overview of the systematic literature review
We performed a systematic review of the literature to identify all published papers that reported the frequency of the USP8 variant and the related pathways in corticotroph pituitary adenomas, detailed clinical presentation and outcomes of patients with and without USP8 mutation and examined the USP8 and EGFR as targeted therapy.
Search strategy
We searched the PubMed, Scopus, web of Sciences, and Cochrane databases. The date of the last search was 20 September 2023 for all databases. We did not apply any language restrictions. Search terms included: “Cushing disease”, “Cushing’s disease”, “Corticotroph adenoma”, “Cushing adenoma”, “Client Cushing disease”, “Atypical corticotroph tumor”, “Corticotroph carcinoma”, “Normal pituitary”, “Corticotroph adenoma”, “Corticotroph Tumor”, “Pituitary ACTH Hypersecretion”, “ACTH-Secreting Pituitary Adenoma”, “Mutation”, “Germline mutation”, “Sporadic mutation”, USP8, “ubiquitin specific peptidase 8”, USP48, “ubiquitin specific peptidase 48”, “Epidermal growth factor”, EGF, “Epidermal growth factor receptor” EGFR, Biomarker.
Inclusion and exclusion criteria
All published papers including original articles, case reports, and case series were included in this systematic review provided that they have reported the frequency of USP8 variant or EGFR expression in corticotroph pituitary adenomas, compared the clinical presentation and outcomes of patients with and without USP8 variant, or examined USP8 or EGFR as treatment targets in CD. Studies applying any type of tissue namely resected human pituitary adenoma tissue, primary cell cultures, cell lines, and transfected cells were included. Articles were excluded if they included different types of pituitary tumors and did not separately analyze corticotroph adenomas, or if they were written in any language other than English.
Results
Case series
Baseline characteristics of the participants
This study included 19 patients of whom 63% (n = 12) were women. They aged between 17 and 65 years. Baseline cortisol ranged between 20 and 43 mic/dl. The ACTH level ranged between 34 and 164 pg/ml. The basal UFC ranged between 316 and 1153 mic/24 h. All patients presented with micro-adenoma except for two patients, one man and one woman (supplementary Table 1).
Frequency of USP8 gene variants
Sanger sequencing of 20 CD tumors revealed two heterozygous pathogenic variants in 2 samples: the 7-bp deletion variant, c.2151_2157delCTCCTCC, p. Ser718GlnfsTer3 was found in one patient; another patient showed the missense variant, c.2159 C > G, p. Pro720Arg (rs672601311) in exon 14. The pathogenic variants were found only in tumor tissue. Targeted sequencing (exon 10) of USP48 did not detect any pathogenic variant. The somatic variations in our study are in the catalytic conserved domain of USP8 protein and lead to disruption of the interaction between USP8 catalytic domain and 14-3-3 protein (Fig. 1).
Fig. 1
Sanger sequencing of pathogenic variants in USP8 hotspot exon. (A, B) bi-directional sequencing of heterozygous missense variant, c.2159 C > G, in tissue sample, (C) A Sanger sequencing chromatogram of the blood sample detected no germline c.2159 C > G mutation. (D) Sanger sequencing chromatograms confirm the presence of heterozygous deletion (c.2151_2157delCTCCTCC) in tissue sample of patient II
All patients achieved biochemical and structural cures after surgery except for one man and one woman who suffered from persistent disease because the tumors were not completely resected due to invasion into the cavernous sinus. They underwent radiotherapy after surgery. These two patients did not show the USP8 variant. Moreover, one man without evidence of the USP8 variant and the two women with the USP8 variant presented with recurrence after initial remission. They presented with micro-adenoma before surgery (supplementary Table 1).
Systematic review
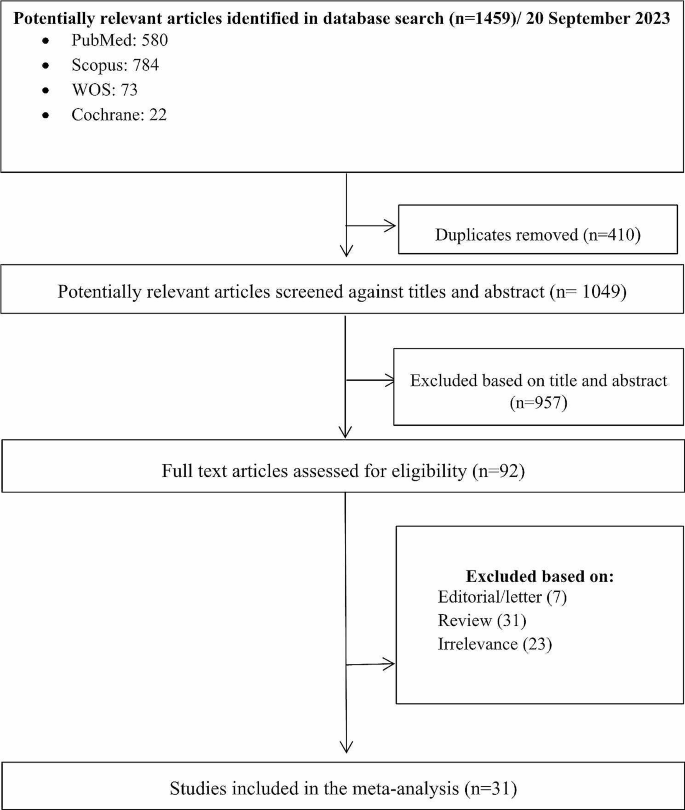
The search yielded 1459 initial results. Upon removing the duplications (n = 410), 1049 studies were reviewed based on the relevancy of their titles and abstracts. Having excluded 957 articles, 92 studies were selected for full-text review. After an in-depth review, 31 articles were selected based on the inclusion and exclusion criteria. A PRISMA diagram detailing the search results is shown in Fig. 2.
Fig. 2
Flow diagram of literature search and study selection
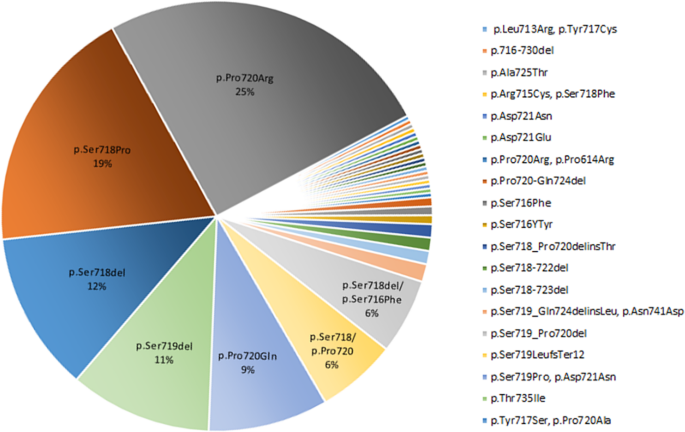
In this systematic review we extracted the information regarding the USP8 variant and the EGFR system in corticotroph adenomas. The USP8 variant was found in 460 individuals with FCPA accounting for 35% of the population included in the related published series (Table 1). Moreover, the highest frequency of missense mutation was found in the 720 code region, p.Pro720Arg (25%), followed by 19% in p.Ser718Pro (Fig. 3). In addition, the frequency of frame-shift and in-frame deletion observed in p.Ser718del and p.Ser719del was 12% and 11%, respectively (Fig. 3).
USP8 variants and the related pathways in corticotroph adenomas
In a study of 42 patients with corticotroph adenomas, USP8 variants were as follows: p. P720R (found in five patients), p. S718P (found in two patients), p. P720Q (found in two patients), p. S716Y (found in one patient), and p. S716F (found in one patient) [12]. Another genetic study demonstrated mutated USP8 deubiquitinating EGFR more effectively than wild type USP8. Some variants namely p.S718del, p.718SP, and p.P720R have higher deubiquitinated activity, while others including p.S718C, p.L713R, and p.Y717C showed similar activity compared to the wild type. These variants have been shown to increase the catalytic and proteolytic activity of USP8, which ultimately leads to the activation of the EGFR pathway. High EGFR levels, in turn, stimulate POMC gene transcription and increase plasma ACTH levels [6]. In the study of Seata, the USP8 variant was found in 23% of corticotroph adenomas. The variants were heterozygous, including p.S718, p.P720 (n = 18), p.S719del (n = 10), and p.P720_723 del (n = 1). Moreover, a comparison of 5 USP8 mutant vs. 34 wild-type specimens indicated different gene expression profile. According to the results, 2 genes involving in EGF signaling, CMTM8 (CKLFlike MARVEL transmembrane domain containing 8) and MAPK15 (mitogen-activated protein kinase 15), were upregulated in USP8 variant carriers [13]. Bujko et al. found USP8 mutation in 31.3% of patients with FCA and silent corticotroph adenomas (SCA). In-frame and missense mutations were p.Ser718del (7 patients), p.Pro720Arg (5 patients), p.Ser718Pro (2 patients) and p.Pro720Gln (one patient). USP8-mutated adenomas showed higher level of POMC, CDC25A, MAPK4 but lower level of CCND2, CDK6, CDKN1B than USP8-wild-type tumors [14].
Another study investigated the molecular pathogenesis of the spectrum of corticotroph adenomas, including CD, SCA, CCA (Crooke cell adenomas), and ACTH-producing carcinoma using whole exome sequencing. The patients with ACTH-producing carcinoma showed the highest number of variants in USP8, EGFR, TP53, AURKA, CDKN1A, and HSD3B1 genes. The USP8 variant was found in c.2159 C > G (p.Pro720Arg) and was positively correlated to the tumor size. However, the USP8 variant was not present in any of the patients with CD [15].
Martins and colleagues conducted a study to investigate the USP8 variant and its contribution to gene expression of cell cycle regulators including P27/CDKN1B, CCNE1, CCND1, CDK2, CDK4, and CDK6 in 32 corticotroph adenoma. They identified variants in certain hotspot exons, namely p.720R (found in five patients), p.S718del (found in three patients), p.S718P (found in one patient), and p.S719_T723del (found in one patient). Moreover, there was no difference regarding the gene expression of the cell cycle regulators CDKN1B (P27), CCNE1 (CYCLIN-E1), CCND1 (CYCLIN-D1), CDK2, CDK4, and CDK6 according to USP8 variant status [16]. Another study investigating the USP8 variants and genes involved in cell cycle regulation observed USP variants including p. P720R (n = 8), p.720Q (n = 2), p. S718SP (n = 2), and an in-frame deletion at the 719 position (n = 8). However, USP8-mutated tumors showed lower CDKN1B, CDK6, CCND2 and higher CDC25A expression. They also observed a significantly lower level of p27 in USP8-mutated tumors as compared to the wild-type ones [17].
A comprehensive study determined the presence of EGFR at the protein and mRNA levels in different pituitary adenomas. The highest incidence of EGFR expression was found in corticotroph adenomas. The corticotroph adenomas with EGFR expression did not show p27 immunoreactivity [18].
DNA methylation regulates promoter activities. The study by Araki et al. identified a novel regulatory region in the human POMC gene which functions as a second promoter. Moreover, they indicated that this region is highly methylated in SCAs and highly demethylated in FCAs and ectopic ACTH-secreting tumors. They also demonstrated demethylation of the second promoter is associated with aggressive features of FCAs independent of the USP8 variant or EGFR signaling. In contrast, the first promoter was highly demethylated in USP8-mutated FCAs [19]. Weigand et al. indicated that p27/kip1 protein expression significantly decreased in USP8-mutated adenomas compared to the wild-type USP8 tumors. Moreover, higher expression of heat shock protein 90 (HSP90) and an increase in the phosphorylation of the transcription factor CREB was observed in mutated-USP8 adenomas [20]. Achaete-scute complex homolog 1 (ASCL1) plays an important role in cell proliferation and also regulates POMC in the cell line. In a recent study, genetic analysis of corticotroph adenomas using RNA-seq and IHC showed an increase in ASCL1expression and protein levels in both mutated and wild type USP8 among CD patients [21].
Genotype-phenotype correlation in USP8-mutated individuals with functional corticotroph adenoma
Sanger sequencing of 120 FCPAs indicated the somatic USP8 variant more frequently in women than in men, which was associated with a significant lower size and higher ACTH level. Moreover, compared to the wild-type tumors, the USP8-mutated ones display a higher level of EGFR expression with a higher staining intensity. The initial remission rate and the recurrence rate in patients initially receiving remission were comparable in both groups [7]. Another study of patients with 134 functional and 11 silent corticotroph adenomas demonstrated somatic USP8 variants only in functional adenomas, none of them occurred in silent adenomas. The USP8 variant in adults was associated with lower age, and predominantly occurred in women. Moreover, the presence of USP8 variant was inversely associated with remission [22]. In a cohort of 42 pediatric patients with FCA, five different USP8 variants (three missenses, one frame-shift, and one in-frame deletion) were identified. None of the patients were found to have gremlin USP8 variants. Patients with somatic USP8 variant were significantly older than those with wild-type USP8. However, there was no significant difference in terms of preoperative hormonal profile and tumor invasiveness between the two groups. However, somatic USP8 mutated patients showed a higher rate of recurrence after a mean follow-up of 34.7 months [23].
In a cohort of 48 FCA, patients with the USP8 variant had significantly higher levels of preoperative urine-free cortisol (UFC). But there was no difference in preoperative ACTH and cortisol level between USP8-mutated and wild-type groups. Although initial remission rate was similar in both groups, patients with USP8 variant revealed a significantly higher rate of recurrence within 10 years follow-up, with a significantly shorter time to recurrence [24]. USP8-mutated FCA patients presented with a significantly larger size of adenoma in a retrospective study. But preoperative hormonal profile and the remission rate were similar in both groups [16]. Retrospective genetic analysis of 92 FCA patients indicated that the USP8 variant was significantly higher in women than men. There was no significant difference in preoperative hormonal profile and tumor size between USP8-mutated and wild-type groups. USP8-mutated carriers were more likely to achieve surgical remission. However, after 10 years follow-up, the recurrence rate was similar in the both groups [25]. A Retrospective study of patients with 30 functional and 20 silent corticotroph adenomas showed USP8 variants in 11 and 2 adenomas, respectively. There was no difference in sex, age, preoperative hormonal profile, and size of the adenomas between patients with and without USP8 variants. However, the USP8-mutated tumors revealed a higher rate of invasiveness. Furthermore, somatostatin receptor 5 (SSRT5) was more frequent in USP8-mutated adenomas [26]. In a retrospective study of FCA patients found no difference considering age at the presentation and hormonal profile between patients with and without USP8 variants. However, macro-adenoma was more frequently seen in USP8-mutated patients. Although initial remission rate was similar in the both groups, after a median 5 (2–8) years of follow-up, USP8-mutated carriers were more likely to develop recurrence [27]. The study conducted by Bujko et al., comparing patients with USP8 mutated and wild-type corticotroph adenomas, demonstrated no difference in age, sex, preoperative hormonal profile, tumor invasiveness, proliferation index, and histology (sparsely vs. densely granulation) between the two groups. However, the USP8-mutated patients showed a higher rate of remission [28].
A cohort of Asian-Indian patients with CD identified that there was no significant difference considering age, sex, tumor size, tumor invasion, and preoperative hormonal profile of the participants with and without the USP8 variant. Moreover, the initial remission rate and long-term recurrence, after a mean follow-up of 25.3 ± 13.6 months, were also comparable in both groups [29]. Liu et al. investigated the expression of EGFR and its signaling pathways in FCAs. They demonstrated that EGFR was overexpressed in 29 of 52 patients with FCA. Moreover, the EGFR signal transducing molecules p-EGFR, p-Akt and p-Erk were upregulated in EGFR-overexpressing adenomas but not in EGFR-negative adenomas. Moreover, the expression of EGFR was positively correlated with ACTH and cortisol levels but not with age, sex, or adenoma size. After a mean follow-up of 42.8 months, 22 patients had tumor recurrence. The EGFR expression was positively associated with the recurrence rate [10].
USP8 and EGFR as potential therapeutic targets in functional corticotroph adenoma
Our systematic search yields nine studies investigating the possible role of the USP8 variant in response to the medications. Four studies evaluated the presence of SSTR5 receptors in USP8– mutated tumors. Genetic analysis of FCAs from a cohort of 39 functional and 23 silent corticotroph adenoma indicated that there was no difference regarding the age of the participants, as well as hormonal profile, size, and invasiveness of the tumor between patients with and without USP8 variants. However, USP8-mutated adenomas showed significantly higher SSRT5 expression compared to the wild-type ones [26].
In a cohort study, USP8-mutated FCA patients were dominantly women and showed lower ACTH levels and smaller tumor size, but no difference in cortisol level. Remission rate was significantly higher in USP8-mutated patients compared to the wild-type ones. Moreover, USP8-mutated adenomas were more likely to express SSTR5 [30]. Genetic analysis of 51 FFPE tumors (21 USP8-mutated and 30 wild-type) indicated significantly higher SSTR5 immunoreactivity score in USP8-mutated tumors, regardless of mutation type. Moreover, in vitro study of 24 corticotroph tumors freshly obtained after TSS indicated a significantly better response to Pasireotide treatment, defined as suppression of ACTH secretion, in human corticotroph tumors carrying USP8 variants [31].
A more recent study aimed to investigate the impact of USP8 variants on in vitro response to Pasirotide in primary cultures obtained from 7 FCAs and also in murine corticotroph tumor cells. USP8 variant in both primary cultured cells and AtT20 cells was associated with higher SSTR5 expression. Moreover, this study indicated although associated with SSTR5 upregulation, mutations at the amino acid 718 of USP8 are not associated with a favorable response to pasireotide, whereas USP8 variants at the amino acid 720 might preserve pasireotide responsiveness [32].
Inhibition of EGFR using Gefitinib, a tyrosine kinase inhibitor, in surgically resected human and canine corticotroph cultured tumors suppressed expression of POMC. Moreover, Blocking EGFR activity in mice attenuated POMC expression, inhibited corticotroph tumor cell proliferation, and induced apoptosis [33]. Araki et al. conducted a study to investigate the utility of EGFR as a therapeutic target for CD. EGFR expression was observed by 2.5 months in transgenic (Tg) mice; and aggressive ACTH-secreting pituitary adenomas with features of Crooke’s cells developed by 8 months with 65% penetrance observed. Moreover, they used the EGFR tyrosine kinase inhibitor Gefitinib to confirm reversibility of EGFR effects on ACTH. Gefitinib suppressed tumor POMC expression and downstream EGFR tumor signaling. Plasma ACTH level and pituitary tumor size was significantly lower in Gefitinib group [34].
Another experimental study investigated the effect of Lapatinib, a potent tyrosine kinase inhibitor, on ACTH production and cell proliferation in AtT-20 mouse corticotroph tumor cells. Lapatinib inhibits EGFR. In this study, Lapatinib decreased proopiomelanocortin (POMC) mRNA levels and ACTH levels in AtT-20 cells and also inhibited cell proliferation and induced apoptosis. Inhibition of EGFR signaling contributes to the inhibition of ACTH production and cell proliferation in corticotroph adenomas [35].
The effect of a potent and selective Jak2 inhibitor, SD1029, on ACTH production and proliferation investigated in mouse AtT20 corticotroph tumor cells. They observed that Jak2 inhibitor SD1029 decreased both POMC transcript levels and basal ACTH levels. These in vitro experiments suggest the Jak2 inhibitor suppresses both the autonomic synthesis and release of ACTH in corticotroph tumor cells. SD1029 was also found to inhibit AtT20-cell proliferation. In addition, SD1029 decreased and increased PTTG1 and GADD45β transcript levels, respectively. They seem to contribute, in part, in the Jak2-induced suppression of cell proliferation and ACTH synthesis [36]. An experimental study examined the effect of USP8 inhibitor on EGFR expression level, and cell viability using AtT20 cells treated with 9-ehtyloxyimino9H-indeno (1, 2-b) pyrazine-2,3-dicarbonitrile, a synthesized USP8 inhibitor. This study demonstrated that treatment with USP8 inhibitor, 9‑ehtyloxyimino9H‑indeno(1,2‑b) pyrazine‑2,3 dicarbonitrile, suppresses ACTH secretion, cell viability, and promotes cell apoptosis in AtT20 cells suggesting that USP8 inhibitor could be a new therapeutic candidate for CD [37].
Kageyama et al. investigated the effects of a potent USP8 inhibitor, DUBs-IN-2, on ACTH production and cell proliferation in mouse corticotroph tumor (AtT-20) cells. DUBs-IN-2 decreased Proopiomelanocortin (POMC) mRNA and ACTH levels. Furthermore, DUBs-IN-2 decreased At-20 cell proliferation and induced apoptosis in corticotroph tumor cells [38]. Another study explored the potential effect of the USP8 inhibitor RA-9 on USP8-WT human tumor corticotroph cells and murine AtT-20 cells. RA-9 significantly decreased cell proliferation and increased cell apoptosis in AtT-20 cells. Moreover, RA-9 reduced ACTH release by USP8-mutant cells. The combined treatment with RA-9 and pasireotide resulted in more efficient in inhibiting ACTH secretion compared with RA-9 or pasireotide alone. Furthermore, similar to pasireotide, RA-9 was able to significantly reduce phospho- ERK1/2 levels in both AtT-20 cells and primary cultured cells from corticotropinomas [39].
Another study, investigating the USP8 variants and genes involved in cell cycle regulation, looked for the role of USP8 variants or a changed p27 level in the response to Palbociclib, Flavopiridol, and Roscovitine, in vitro, using murine corticotroph AtT-20/D16v-F2 cells. They did not found any significant difference in cell viability or cell proliferation between the AtT-20/D16v-F2 cells overexpressing wild-type and mutated USP8 that were treated with cell cycle inhibitors. There was also no difference in the response to inhibitors of CKDs in the cells with overexpression of p27 and control cells [17].
Analytical conclusion
In our series, we found two USP8 variants including a 7-bp deletion variant, c.2151_2157delCTCCTCC, p. Ser718GlnfsTer3, and a missense variant, c.2159 C > G, p. Pro720Arg (rs672601311) in exon 14. Moreover, the systematic review of the published data indicated that 35% of corticotroph adenomas harbor USP8 variant the most of which was found in the 720 code region, p. Pro720Arg. Similar to the most previous studies, the USP-8 mutated patients were women, presented with micro-adenoma and experienced recurrence after initial remission.
We systematically reviewed the literature regarding the USP8 variant in corticotroph adenomas and classified the results into three categories; including USP8 variants and the related pathways, genotype-phenotype correlation in USP8-mutated individuals, and USP8 and EGFR as potential therapeutic targets.
Different USP8 variants are identified in corticotroph adenomas. Activation of the EGFR pathway is a well-established consequence of USP8 variants [6, 15]. But there is inconsistency regarding the role of USP8 variants in cell cycle regulation in corticotroph adenomas. Some studies showed no difference in the gene expression of the cell cycle regulators CDKN1B (P27), CCNE1 (CYCLIN-E1), CCND1 (CYCLIN-D1), CDK2, CDK4, and CDK6 according to USP8 variant status [21]; while the others indicated USP8-mutated tumors have lower CDKN1B, CDK6, CCND2 and higher CDC25A expression [20]. Moreover, demethylation of the first promoter is affected with USP8 variant status [19]. However, more studies are required to establish the pathway underlying the USP8 variants.
Data regarding sex, age, hormonal level, tumor size, and clinical outcomes in USP8-mutated individuals with FCA are relatively consistent among different studies. The USP8 variant seems to be associated with younger age and is more likely to occur in women. Meta-analysis of data from ten series indicated USP8 variant is 2.63 times higher in women than in men [40]. Since CD is more prevalent in young women, the potential effect of estrogen on the growth of USP8-mutant corticotroph cells has been hypothesized. There is evidence that corticotroph cells express estrogen receptors [41]. Moreover, in vitro studies indicated estrogen can stimulate corticotroph cell proliferation mediated by EGFR signaling pathways [42]. More precise studies are required to better explain the age-sex distribution of USP8 variant in patients with CD.
Results regarding the hormonal pattern among the series are partly controversial. Two series indicated significantly higher levels of ACTH and UFC in USP8-mutated patients compared to the wild-type ones [7, 24]. Moreover, one study demonstrated the expression levels of EGFR were positively correlated with ACTH and cortisol levels [10]. Conversely, one study showed a significantly lower ACTH level in patients with the USP8 variant [30]. However, in a systematic analysis of the two series the correlation of UFC and USP8 variant did not reach a significant difference, this might be due to the small number of cases included in the analysis [40].
There are also some discrepancies on tumor size and invasiveness in USP8-mutated tumors. Some studies indicated a significant smaller size in USP8-mutated tumors, while others showed a significant larger size in USP8-mutated tumors. But some study found no significant difference regarding tumor size and invasiveness between USP8-mutated and wild-type tumors. A recent systematic analysis of magnetic resonance imaging (MRI) findings from individuals with CD indicated USP8-mutated tumors are more likely to be less than 10 mm compared to wild-type ones [40]. Moreover, a cohort of 60 patients with FCA indicated smaller tumor size and less invasiveness in USP8-mutated tumors [30]. In contrast to these findings, a cohort of Brazilian patients observed a tendency toward more somatic USP8 variant in tumors more than 10 mm in size [40]. These discrepancies might be due to the different methods used for extraction of MRI data.
Considering the clinical outcomes, most studies indicated a higher remission rate except for one that showed a significantly lower rate of remission in USP8-mutated patients [22, 25, 28, 30]. Moreover, some studies demonstrated a higher rate of recurrence in carriers of USP8 variant [24, 27, 42]. However, other studies found no significant difference neither in the initial remission nor in the late recurrence rate between the carriers of USP8 variant and the individuals with wild-type USP8. The inconsistency in the results might be due to the lack of a systematic protocol for evaluation of these patients. Moreover, the number of patients included in the different studies was relatively low. Further multicenter prospective studies with the same protocol are required to yield more consistent information regarding the influence of USP8 variant on the clinical presentation as well as early and late outcomes of FCAs.
There are promising studies regarding USP8-targeted therapy. We found evidence that USP8-mutated tumors have higher SSRT5 expression [30, 31]. Moreover, in vitro studies demonstrated that Pasirotide suppressed ACTH secretion significantly more in the USP8-mutated tumors than in wild-type ones [31]. These evidences suggest that USP8 mutational status could be used as a marker of Pasirotide response in CD. Furthermore, USP8-mutated tumors are more likely to express EGFRs compared to the wild-type ones [6]. Inhibition of EGFR using Gefitinib and Lapatinib has been associated with promising results regarding the EGFR-targeting therapy in CD [33,34,35]. Moreover, experimental studies of two USP8 inhibitors, 9‑ehtyloxyimino9H‑indeno (1,2‑b) pyrazine‑2,3 dicarbonitrile and DUBs-IN-2, have shown their potential to suppress POMC expression and ACTH secretion, decrease cell proliferation, and promote apoptosis [37, 38].
In summary, the studies investigated the association of USP8– variants and clinical manifestations as well as clinical outcomes of the corticotroph adenomas are partly inconsistent. More precise multicenter studies are required to yield more consistent information regarding the phenotype-genotype correlation and to develop effective targeted therapies.
Data availability
The datasets used and/or analyzed during the current atudy are available from the corresponding author on reasonable request.
Abbreviations
ABI:
Applied Biosystems
ACTH:
Adrenocorticotropic Hormone
CCA:
Crooke Cell Adenomas
CD:
Cushing’s Disease
DNA:
Deoxyribonucleic Acid
EGFR:
Epidermal Growth Factor Receptor
Erk:
Extracellular Signal-Regulated Kinases
FCAs:
Functional Corticotroph Adenomas
FCPA:
Functional Corticotroph Pituitary Adenoma
FFPE:
Formalin-Fixed And Paraffin-Embedded
ICH-GCP:
International Conference On Harmonization Of Good Clinical Practice
IHC:
Immunohistochemistry
MRI:
Magnetic Resonance Imaging
PCR:
Polymerase Chain Reaction
PRISMA:
Preferred Reporting Items For Systematic Reviews And Meta-Analyses
RNA-seq:
RNA Sequencing
SCA:
Silent Corticotroph Adenomas
TSS:
Transsphenoidal Surgery
USP8:
Ubiquitin-Specific Peptidase
USP48:
Ubiquitin Specific Peptidase 48
References
Uwaifo GI, Hura DE. Hypercortisolism. [Updated 2023 Jul 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. https://www.ncbi.nlm.nih.gov/books/NBK551526/.
Pivonello R, Simeoli C, Di Paola N, Colao A. Cushing’s disease: adrenal steroidogenesis inhibitors. Pituitary. 2022;25(5):726–32.
Balomenaki M, Vassiliadi DA, Tsagarakis S. Cushing’s disease: risk of recurrence following trans-sphenoidal surgery, timing and methods for evaluation. Pituitary. 2022;25(5):718–21.
Seltzer J, Ashton CE, Scotton TC, Pangal D, Carmichael JD, Zada G. Gene and protein expression in pituitary corticotroph adenomas: a systematic review of the literature. Neurosurg Focus. 2015;38(2):E17.
Reincke M, Sbiera S, Hayakawa A, Theodoropoulou M, Osswald A, Beuschlein F, Meitinger T, Mizuno-Yamasaki E, Kawaguchi K, Saeki Y, et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat Genet. 2015;47(1):31–8.
Ma ZY, Song ZJ, Chen JH, Wang YF, Li SQ, Zhou LF, Mao Y, Li YM, Hu RG, Zhang ZY, et al. Gain-of-function USP8 mutations in Cushing’s disease. Cell Res. 2015;25(3):306–17.
Naviglio S, Mattecucci C, Matoskova B, Nagase T, Nomura N, Di Fiore PP, Draetta GF. UBPY: a growth-regulated human ubiquitin isopeptidase. EMBO J. 1998;17(12):3241–50.
Liu X, Feng M, Dai C, Bao X, Deng K, Yao Y, Wang R. Expression of EGFR in Pituitary Corticotroph Adenomas and its RelationshipWith Tumor Behavior. Front Endocrinol (Lausanne). 2019;10:785.
Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, Montori VM. The diagnosis of Cushing’s syndrome: an endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93(5):1526–40.
Ballmann C, Thiel A, Korah HE, Reis AC, Saeger W, Stepanow S, Köhrer K, Reifenberger G, Knobbe-Thomsen CB, Knappe UJ, Scholl UI. USP8 Mutations in Pituitary Cushing Adenomas-Targeted Analysis by Next-Generation Sequencing. J Endocr Soc. 2018;2(3):266–278.
Sesta A, Cassarino MF, Terreni M, Ambrogio AG, Libera L, Bardelli D, et al. Ubiquitin-specific protease 8 mutant corticotrope Adenomas Present Unique secretory and molecular features and shed light on the role of Ubiquitylation on ACTH Processing. Neuroendocrinology. 2020;110(1–2):119–29.
Bujko M, Kober P, Boresowicz J, Rusetska N, Paziewska A, Dąbrowska M, Piaścik A, Pękul M, Zieliński G, Kunicki J, et al. USP8 mutations in corticotroph adenomas determine a distinct gene expression profile irrespective of functional tumour status. Eur J Endocrinol. 2019;181(6):615–27.
Andonegui-Elguera S, Silva-Román G, Peña-Martínez E, Taniguchi-Ponciano K, Vela-Patiño S, Remba-Shapiro I, Gómez-Apo E, Espinosa-de-Los-Monteros AL, Portocarrero-Ortiz LA, Guinto G, et al. The genomic Landscape of Corticotroph tumors: from Silent Adenomas to ACTH-Secreting Carcinomas. Int J Mol Sci. 2022;23(9):4861.
Mossakowska BJ, Rusetska N, Konopinski R, Kober P, Maksymowicz M, Pekul M, Zieliński G, Styk A, Kunicki J, Bujko M. The expression of cell cycle-related genes in USP8-Mutated corticotroph neuroendocrine pituitary tumors and their possible role in cell cycle-targeting treatment. Cancers (Basel). 2022;14(22):5594.
Theodoropoulou M, Arzberger T, Gruebler Y, Jaffrain-Rea ML, Schlegel J, Schaaf L, Petrangeli E, Losa M, Stalla GK, Pagotto U. Expression of epidermal growth factor receptor in neoplastic pituitary cells: evidence for a role in corticotropinoma cells. J Endocrinol. 2004;183(2):385–94.
Araki T, Tone Y, Yamamoto M, Kameda H, Ben-Shlomo A, Yamada S, Takeshita A, Yamamoto M, Kawakami Y, Tone M, Melmed S. Two distinctive POMC promoters modify Gene expression in Cushing Disease. J Clin Endocrinol Metab. 2021;106(9):e3346–63.
Weigand I, Knobloch L, Flitsch J, Saeger W, Monoranu CM, Höfner K, Herterich S, Rotermund R, Ronchi CL, Buchfelder M, et al. Impact of USP8 gene mutations on protein deregulation in Cushing Disease. J Clin Endocrinol Metab. 2019;104(7):2535–46.
Chen Z, Jia Q, Zhao Z, Zhang Q, Chen Y, Qiao N, Ye Z, Ji C, Zhang Y, He W, et al. Transcription factor ASCL1 acts as a novel potential therapeutic target for the treatment of the Cushing’s Disease. J Clin Endocrinol Metab. 2022;107(8):2296–306.
Perez-Rivas LG, Theodoropoulou M, Ferraù F, Nusser C, Kawaguchi K, Stratakis CA, Faucz FR, Wildemberg LE, Assié G, Beschorner R, et al. The gene of the ubiquitin-specific protease 8 is frequently mutated in Adenomas causing Cushing’s Disease. J Clin Endocrinol Metab. 2015;100(7):E997–1004.
Faucz FR, Tirosh A, Tatsi C, Berthon A, Hernández-Ramírez LC, Settas N, Angelousi A, Correa R, Papadakis GZ, Chittiboina P, et al. Somatic USP8 gene mutations are a Common cause of Pediatric Cushing Disease. J Clin Endocrinol Metab. 2017;102(8):2836–43.
Albani A, Pérez-Rivas LG, Dimopoulou C, Zopp S, Colón-Bolea P, Roeber S, Honegger J, Flitsch J, Rachinger W, Buchfelder M. The USP8 mutational status may predict long-term remission in patients with Cushing’s disease. Clin Endocrinol (Oxf). 2018.
Losa M, Mortini P, Pagnano A, Detomas M, Cassarino MF, Pecori Giraldi F. Clinical characteristics and surgical outcome in USP8-mutated human adrenocorticotropic hormone-secreting pituitary adenomas. Endocrine. 2019;63(2):240–6.
Castellnou S, Vasiljevic A, Lapras V, Raverot V, Alix E, Borson-Chazot F, Jouanneau E, Raverot G, Lasolle H. SST5 expression and USP8 mutation in functioning and silent corticotroph pituitary tumors. Endocr Connect. 2020;9(3):243–53.
Treppiedi D, Barbieri AM, Di Muro G, Marra G, Mangili F, Catalano R, Esposito E, Ferrante E, Serban AL, Locatelli M, et al. Genetic profiling of a cohort of Italian patients with ACTH-Secreting pituitary tumors and characterization of a novel USP8 gene variant. Cancers (Basel). 2021;13(16):4022.
Bujko M, Kober P, Boresowicz J, Rusetska N, Zeber-Lubecka N, Paziewska A, Pekul M, Zielinski G, Styk A, Kunicki J, Ostrowski J, et al. Differential microRNA expression in USP8-Mutated and wild-type Corticotroph Pituitary tumors reflect the difference in protein ubiquitination processes. J Clin Med. 2021;10(3):375.
Abraham AP, Pai R, Beno DL, Chacko G, Asha HS, Rajaratnam S, Kapoor N, Thomas N, Chacko AG. USP8, USP48, and BRAF mutations differ in their genotype-phenotype correlation in Asian Indian patients with Cushing’s disease. Endocrine. 2022;75(2):549–59.
Hayashi K, Inoshita N, Kawaguchi K, Ibrahim Ardisasmita A, Suzuki H, Fukuhara N, Okada M, Nishioka H, Takeuchi Y, Komada M, et al. The USP8 mutational status may predict drug susceptibility in corticotroph adenomas of Cushing’s disease. Eur J Endocrinol. 2016;174(2):213–26.
Treppiedi D, Marra G, Di Muro G, Esposito E, Barbieri AM, Catalano R, Mangili F, Bravi F, Locatelli M, Lania AG, et al. P720R USP8 mutation is Associated with a better responsiveness to Pasireotide in ACTH-Secreting PitNETs. Cancers. 2022;14(10):2455.
Fukuoka H, Cooper O, Ben-Shlomo A, Mamelak A, Ren SG, Bruyette D, Melmed S. EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. J Clin Invest. 2011;121(12):4712–21.
Araki T, Liu X, Kameda H, Tone Y, Fukuoka H, Tone M, Melmed S. EGFR Induces E2F1-Mediated Corticotroph Tumorigenesis, J Endocr Soc. 2017; 2017;1(2):127–143.
Asari Y, Kageyama K, Sugiyama A, Kogawa H, Niioka K, Daimon M. Lapatinib decreases the ACTH production and proliferation of corticotroph tumor cells. Endocr J. 2019;66(6):515–22.
Asari Y, Kageyama K, Nakada Y, Tasso M, Takayasu S, Niioka K, Ishigame N, Daimon M. Inhibitory effects of a selective Jak2 inhibitor on adrenocorticotropic hormone production and proliferation of corticotroph tumor AtT20 cells. Onco Targets Ther. 2017;10:4329–38.
Jian FF, Li YF, Chen YF, Jiang H, Chen X, Zheng LL, Zhao Y, Wang WQ, Ning G, Bian LG, Sun QF. Inhibition of Ubiquitin-specific peptidase 8 suppresses adrenocorticotropic hormone production and tumorous corticotroph cell growth in AtT20 cells. Chin Med J. 2016;129(17):2102–8.
Kageyama K, Asari Y, Sugimoto Y, Niioka K, Daimon M. Ubiquitin-specific protease 8 inhibitor suppresses adrenocorticotropic hormone production and corticotroph tumor cell proliferation. Endocr J. 2020;67(2):177–84.
Wanichi IQ, de Paula Mariani BM, Frassetto FP, Siqueira SAC, de Castro Musolino NR, Cunha-Neto MBC, Ochman G, Cescato VAS, Machado MC, Trarbach EB, et al. Cushing’s disease due to somatic USP8 mutations: a systematic review and meta-analysis. Pituitary. 2019;22(4):435–42.
Chaidarun SS, Swearingen B, Alexander JM. Differential expression of estrogen receptor-beta (ER beta) in human pituitary tumors: functional interactions with ER alpha and a tumor-specific splice variant. J Clin Endocrinol Metab. 1998;83(9):3308–15.
We thank all the participants enrolled in this study.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Iran University of Medical Sciences No. IR.IUMS.REC.1398.082.
Author information
Author notes
Nahid Hashemi-Madani and Sara Cheraghi are joint first authors.
Authors and Affiliations
Endocrine Research Center, Institute of Endocrinology and Metabolism, Iran University of Medical Sciences, Tehran, Iran, No. 10, Firoozeh St., Vali-asr Ave., Vali-asr Sq, Tehran, Iran
Nahid Hashemi-Madani, Sara Cheraghi, Zahra Emami & Mohammad E. Khamseh
Department of Pathology, Firoozgar hospital, Iran University of Medical Sciences, Tehran, Iran
Ali Zare Mehrjardi
Department of Endocrinology, Arad Hospital, Tehran, Iran
Mahmoud Reza Kaynama
Contributions
Conception and design: NHM and MEK; Development of methodology: NHM, SC and ZE; Acquisition, analysis, and interpretation of data: NHM, SC, ZE and AZM; Writing, review, and/or revision of the manuscript: NHM, SC, ZE, MRK and MEK; Administrative, technical or, material support: NHM, MEK; Study supervision: MEK; All authors read and approved the final manuscript.
This study was performed in accordance with the 1964 Helsinki Declaration, and was approved by the Ethics Committee of Iran University of Medical Sciences. Informed consent was obtained from all individual participants included in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Additional file 1 of Targeted analysis of Ubiquitin-Specific Peptidase (USP8) in a population of Iranian people with Cushing’s disease and a systematic review of the literature
Skip to figshare navigation
Below is the link to the electronic supplementary material.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Hashemi-Madani, N., Cheraghi, S., Emami, Z. et al. Targeted analysis of Ubiquitin-Specific Peptidase (USP8) in a population of Iranian people with Cushing’s disease and a systematic review of the literature. BMC Endocr Disord24, 86 (2024). https://doi.org/10.1186/s12902-024-01619-z
There is increasing evidence that multisystem morbidity in patients with Cushing’s disease (CD) is only partially reversible following treatment. We investigated complications from multiple organs in hospitalized patients with CD compared to patients with non-functioning pituitary adenoma (NFPA) after pituitary surgery.
Design
Population-based retrospective cohort study using data from the Swiss Federal Statistical Office between January 2012 and December 2021.
Methods
Through 1:5 propensity score matching, we compared hospitalized patients undergoing pituitary surgery for CD or NFPA, addressing demographic differences. The primary composite endpoint included all-cause mortality, major adverse cardiac events (i.e., myocardial infarction, unstable angina, heart failure, cardiac arrest, ischemic stroke), hospitalization for psychiatric disorders, sepsis, severe thromboembolic events, and fractures in need of hospitalization. Secondary endpoints comprised individual components of the primary endpoint and surgical reintervention due to disease persistence or recurrence.
Results
After matching, 116 patients with CD (mean age 45.4 years [SD, 14.4], 75.0% female) and 396 with NFPA (47.3 years [14.3], 69.7% female) were included and followed for a median time of 50.0 months (IQR 23.5, 82.0) after pituitary surgery. CD presence was associated with a higher incidence rate of the primary endpoint (40.6 vs. 15.7 events per 1,000 person-years, HR 2.75; 95% CI, 1.54 to 4.90). CD patients also showed increased hospitalization rates for psychiatric disorders (HR 3.27; 95% CI, 1.59 to 6.71) and a trend for sepsis (HR 3.15; 95% CI, 0.95 to 10.40).
Conclusions
Even after pituitary surgery, CD patients faced a higher hazard of complications, especially psychiatric hospitalizations and sepsis.
Accepted manuscripts are PDF versions of the author’s final manuscript, as accepted for publication by the journal but prior to copyediting or typesetting. They can be cited using the author(s), article title, journal title, year of online publication, and DOI. They will be replaced by the final typeset articles, which may therefore contain changes. The DOI will remain the same throughout.
Cushing’s syndrome, a clinical condition characterized by hypercortisolemia, exhibits distinct clinical signs and is associated with cyclic cortisol secretion in some patients. The clinical presentation of cyclic Cushing’s syndrome can be ambiguous and its diagnosis is often challenging.
We experienced a 72-year-old woman with cyclic ACTH-dependent Cushing’s syndrome caused by a pulmonary carcinoid tumor. Diagnosis was challenging because of the extended trough periods, and the responsible lesion was initially unidentified. A subsequent follow-up computed tomography revealed a pulmonary lesion, and ectopic ACTH secretion from this lesion was confirmed by pulmonary artery sampling. Despite the short peak secretion period of ACTH (approximately one week), immunostaining of the surgically removed tumor confirmed ACTH positivity. Interestingly, stored plasma chromogranin A levels were elevated during both peak and trough periods.
The experience in evaluating this patient prompted us to investigate the potential use of plasma chromogranin A as a diagnostic marker of ACTH-dependent Cushing’s syndrome. A retrospective study was conducted to determine the efficacy of plasma chromogranin A in three patients with ectopic ACTH syndrome (EAS), including the present case, and six patients with Cushing’s disease (CD) who visited our hospital between 2018 and 2021. Notably, plasma chromogranin A levels were higher in patients with EAS than in those with CD. Additionally, a chromogranin A level in the present case during the trough phase was lower than that in the peak phase, and was similar to those in CD patients. The measurement of plasma chromogranin A levels could aid in differentiating EAS from CD.
Spontaneous remission of Cushing’s disease (CD) is uncommon and often attributed to pituitary tumor apoplexy. We present a case involving a 14-year-old female who exhibited clinical features of Cushing’s syndrome. Initial diagnostic tests indicated CD: elevated 24h urinary cortisol (235 µg/24h, n < 90 µg/24h), abnormal 1 mg dexamethasone overnight test (cortisol after 1 mg dex 3.4 µg/dL, n < 1.8 µg/dL), and elevated adrenocorticotropic hormone concentrations (83.5 pg/mL, n 10-60 pg/mL). A pituitary adenoma was suspected, so a nuclear MRI was performed, with findings suggestive of a pituitary microadenoma. The patient was referred for a transsphenoidal resection of the microadenoma. While waiting for surgery, the patient presented to the emergency department with a headache and clinical signs of meningism. A computed axial tomography of the central nervous system was performed, and no structural alterations were found. The symptoms subsided with analgesia. One month later, she presented again to the emergency department with clinical findings of acute adrenal insufficiency (cortisol level of 4.06 µg/dL), and she was noted to have spontaneous biochemical remission associated with the resolution of her symptoms of hypercortisolism. For that reason, spontaneous CD remission induced by pituitary apoplexy (PA) was diagnosed. The patient has been managed conservatively since the diagnosis and remains in clinical and biochemical remission until the present time, after 10 months of follow-up. There are three unique aspects of our case: the early age of onset of symptoms, the spontaneous remission of CD due to PA, which has been rarely reported in the medical literature, and the fact that the patient presented a microadenoma because there are fewer than 10 clinical case reports of PA associated with microadenoma.
Introduction
Cushing’s disease (CD) is characterized by excessive production of adrenocorticotropic hormone by a pituitary adenoma and represents the most common cause of endogenous Cushing’s syndrome (CS) [1]. CD was first reported in 1912 by Harvey Williams Cushing, and he described 12 cases at the Peter Bent Brigham Hospital in Baltimore [2]. This disease has a global incidence of approximately 2.2 cases per 1,000,000 people and occurs more frequently in women from 20 to 50 years of age [3]. Pituitary apoplexy (PA) is a rare condition that occurs in 2-12% of cases, and it has a high morbidity and mortality rate [4]. We report an interesting case of a woman diagnosed with CD who achieved spontaneous remission of her disease after a PA.
Case Presentation
A 14-year-old female presented with a two-year history of weight gain (32 kg), depression, elevated blood pressure, type 2 diabetes mellitus, and growth failure (height less than the third percentile). Her height was 140 cm, and her BMI was 28.1 (97th percentile). At presentation, she had not yet reached menarche. Physical examination revealed Tanner 2 breast development, acne, hirsutism, moon facies, dorsocervical fat pad, central obesity, and stretch marks. Initial laboratory tests showed hemoglobin A1C of 13%, low-density lipoprotein of 167 mg/dL, triglycerides of 344 mg/dL, high-density lipoprotein of 26 mg/dL, creatinine of 0.4 mg/dL, and elevated liver enzymes. Abdominal ultrasound indicated moderate hepatic steatosis changes.
Given the high suspicion of CS, a hormonal profile was conducted (Table 1), confirming CS and subsequently diagnosing CD. A nuclear MRI revealed a 2.6 × 1.8 mm pituitary lesion (Figure 1), prompting referral for transsphenoidal resection of the pituitary microadenoma.
Laboratories
Reference range
Initial
One month
Three months
Six months
TSH (mUI/L)
0.35-4.94
–
2.17
–
2.01
AM cortisol (µg/dL)
6.02-18.4
17.3
4.06
<0.5
4.7
1 mg DST (µg/dL)
<1.8
3.4
–
–
–
8 mg DST (µg/dL)
<50% suppression
1.9 (78% suppression)
–
–
–
Urine-free cortisol (µg/24h)
<90
235
–
–
–
ACTH (pg/mL)
10-60
83.5
–
19.2
9.7
IGF-1 (ng/mL)
36-300
–
–
–
293
Table 1: Pertinent laboratory investigation at baseline and follow-up with our patient
Figure 1: Axial view of a T1 MRI with contrast showing a sellar lesion
The red arrow shows a microadenoma in relation to the normal pituitary gland.
Approximately one month after the suppression tests and while awaiting surgery, the patient presented to the emergency department with a sudden, severe, holocranial headache accompanied by projectile vomiting and diplopia, suggestive of meningism. A computed axial tomography of the central nervous system was conducted, revealing no structural abnormalities. Symptoms resolved with intravenous analgesia within approximately four to six hours. Subsequently, the patient experienced a significant decrease in insulin requirements, ultimately leading to the suspension of insulin therapy due to persistent hypoglycemia.
Weeks after the headache episode, the patient was reevaluated in the emergency department with a three-day history of diffuse abdominal pain, vomiting, asthenia, myalgia, hypotension, tachycardia, orthostatism, and recurrent hypoglycemia despite insulin suspension. Acute adrenal insufficiency was suspected and confirmed by a cortisol level of 4.06 µg/dL. Treatment with intravenous hydrocortisone 50 mg every six hours was initiated, leading to complete resolution of symptoms within 72 hours. The patient was discharged on maintenance therapy with oral hydrocortisone (20 mg in the morning and 10 mg at night). Subsequent follow-ups showed undetectable cortisol levels. Currently, the patient has been followed up for 10 months post-event, showing persistent clinical and hormonal remission of her disease.
Discussion
CD represents approximately 80% of cases of endogenous hypercortisolism, and pituitary microadenomas are the most common cause of CD in all age groups [5]. CD prevalence is 0.3-6.2 cases per 100,000 people [3], which represents 4.4% of all pituitary adenomas [6], and it is up to five times more likely to occur in women than men. Spontaneous remission of CD is rare, and it is mainly due to the apoplexy of a pituitary tumor [7].
PA is a potentially fatal condition resulting from hemorrhage or necrosis of a pituitary adenoma that produces compression of the surrounding structures with symptoms that can be critical and even fatal [8]. PA affects between 2% and 12% of patients with pituitary adenomas, mainly in nonfunctional macroadenomas [9]. Although the main mechanism of PA is hemorrhage, it can also be due to a hemorrhagic infarction or an infarction without hemorrhage; this last scenario is clinically less aggressive [10]. Among the most important precipitating factors are craniocerebral trauma, pregnancy, thrombocytopenia, coagulopathies, pituitary stimulation tests, drugs such as anticoagulants and estrogens, surgeries that are complicated by hypotension, and radiotherapy [4,11,12].
There are three unique aspects of our case. First, the age of onset is 14 years old. This characteristic has been reported in less than 6% of cases of CD, with a mean age of onset between 12.3 and 14.1 years and a slightly higher incidence in men (63%) [13]. In this population, CD is the most common cause of hypercortisolism, accounting for 75-80% of all cases [14]. Furthermore, our patient presented a significant weight gain, severe compromise in her height, hypertension, depression, and diabetes mellitus, which is compatible with the classic profile described for CD in pediatric ages. It is important to clarify that although type 2 diabetes mellitus is common in adults, it is unusual in the pediatric population [13].
Second, spontaneous remission in CD due to apoplexy has been rarely reported in the past; hence, our case is an important addition to the scant literature on this unusual phenomenon. Although there are characteristics suggestive of PA, such as hyperdense lesions within the pituitary gland and the reinforcing ring, a CT scan has a low sensitivity for detecting pituitary hemorrhage (21-46%); therefore, a negative CT scan does not rule out PA in cases where there is infarction without hemorrhage, a situation that could correspond to our case [15].
The third unique feature of our case is that the stroke occurred in the context of a microadenoma, a situation reported in less than 10 cases in the literature. Despite being a microadenoma, the symptoms of PA were severe, with symptoms of meningism, an intense headache, vomiting, and the development of adrenal insufficiency. Taylor et al. [16] reported a similar case of a 41-year-old female with microadenoma whose PA was associated with severe headache and vomiting.
The main differential diagnosis in our case is cyclical CS (CCS), a disorder that occurs in 15% of CS cases, especially in CD [17]. The diagnosis of CCS is classically established with three peaks and two valleys in cortisol secretion, spontaneous fluctuations, and clinical features of CS [7]. The possibility of CCS was ruled out due to the typical presentation of the PA event and the persistence of hypocortisolism.
Finally, several cases of recurrence of their disease have been described after remission of CS due to AP. Those recurrences usually develop in follow-ups of up to seven years [18]. At the time of the last evaluation (10 months post-PA), the patient remained in remission, but long-term follow-up is required to detect both reactivation and hypopituitarism [19].
Conclusions
CD is a rare entity in the pediatric population, usually associated with a pituitary microadenoma. Spontaneous remission of this disease is very uncommon, but when it occurs, it is mainly due to PA. We describe a case with three unique aspects: CD with an early age of onset of symptoms, spontaneous remission of CD due to PA, which has been rarely reported in the medical literature, and the fact that there are less than 10 clinical case reports of PA associated with microadenoma. It is imperative for clinicians to be aware of this possible outcome in patients with CD.