


Journal of the Endocrine Society, Volume 6, Issue 7, July 2022, bvac080, https://doi.org/10.1210/jendso/bvac080
Abstract
Subclinical pituitary hemorrhage, necrosis, and/or cystic degeneration (SPH) presents mainly in large tumors and prolactinomas. The characteristics of patients with Cushing disease (CD) and SPH are not known.
To determine if SPH affects the presentation and biochemical profile of young patients with CD.
Pediatric and adolescent patients who were diagnosed with CD between 2005 and 2021 and available magnetic resonance imaging images were evaluated for SPH. The clinical and biochemical characteristics of patients with and without SPH were compared.
Evidence of possible SPH was present in 12 out of 170 imaging studies (7.1%). Patients with and without SPH had similar age at diagnosis and sex distribution but differed in disease duration (median duration: 1.0 year [1.0-2.0] in the SPH group vs 2.5 years [1.5-3.0] in the non-SPH group, P = .014). When comparing their biochemical evaluation, patients with SPH had higher levels of morning adrenocorticotropin (ACTH) (60.8 pg/mL [43.5-80.3]) compared to patients without SPH (39.4 pg/mL [28.2-53.2], P = .016) and the degree of cortisol reduction after overnight high dose (8 mg or weight-based equivalent) dexamethasone was lower (–58.0% [–85.4 to –49.7]) compared to patients without SPH (85.8 [–90.5 to –76.8], P = .035). The presence of SPH did not affect the odds of remission after surgery or the risk of recurrence after initial remission.
SPH in ACTH-secreting pituitary adenomas may affect their biochemical response during endocrine evaluations. They may, for example, fail to suppress to dexamethasone which can complicate diagnosis. Thus, SPH should be mentioned on imaging and taken into consideration in the work up of pediatric patients with CD.
Acute hemorrhage or necrosis of pituitary adenomas (PAs), defined as pituitary apoplexy, is a rare life-threatening condition that requires emergent neurosurgical evaluation [1]. However, subclinical hemorrhage, necrosis, intratumor cystic degeneration, and/or infarct of PAs, herein all events included in the term subclinical pituitary hemorrhage (SPH) for brevity, may occur in up to 7% to 22% of all pituitary tumors [2-9]. SPH is not associated with significant clinical symptoms and is often discovered at the time of routine diagnostic evaluation [2-9].
Previous studies suggested that SPH is more common in large tumors, prolactin-secreting or nonfunctioning PAs, while other factors, such as initiation or withdrawal of treatment with dopamine agonists, use of anticoagulants and others, have also been hypothesized to be involved in this process [5, 6]. Overall, adrenocorticotropin (ACTH)-secreting PAs represent small percentage of SPH (0%-3.2% of cases reported) [3, 5, 6, 8]. Although pituitary apoplexy is associated with pituitary hormone deficiencies, SPH has a lower if any effect on the function of the remaining pituitary gland when it occurs in nonfunctioning adenomas [3, 4].
The diagnosis of Cushing syndrome (CS) involves elaborate and time-dependent tests that are based on cortisol secretion and its regulation by ACTH [10]. Furthermore, the differential diagnosis of ACTH-dependent causes between ACTH-secreting PAs (Cushing disease, CD) and ectopic ACTH secretion is based on several dynamic tests, such as corticotropin-releasing hormone (CRH) stimulation and dexamethasone suppression [11]. The biochemical profile of corticotropinomas with SPH to both baseline and dynamic endocrine tests is not known.
Materials and Methods
Participants
Individuals enrolled under the protocol 97-CH-0076 (ClinicalTrials.gov identifier NCT00001595) at the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), from 2005 to 2020 with confirmed diagnosis of CD, were screened for eligibility in the study. Pediatric and adolescent patients (diagnosis age < 21 years) with imaging studies available before any surgical intervention were included in the analysis. Patients with previous surgery of the pituitary gland or who were evaluated during recurrence were excluded from the study since postoperative changes make imaging findings difficult to distinguish from true SPH, and biochemical presentation at recurrence often differs in severity from initial diagnosis. CS diagnosis was based on criteria defined by the Endocrine Society guidelines and adjusted for the pediatric and adolescent population as needed (abnormal measurements in at least 2 of the following criteria: elevated 24 hour urinary free cortisol [UFC], elevated midnight serum cortisol [> 4.4 mcg/dL in children or > 7.5 mcg/dL for patients age > 18 years], and/or failure to suppress cortisol to 1 mg [or weight-based equivalent dose] overnight dexamethasone suppression test [postdexamethasone cortisol > 1.8 mcg/dL]). CD diagnosis was based on postoperative histologic confirmation of ACTH-secreting PA in most cases, or remission after pituitary surgery even if histologic report failed to identify a PA in the studied material. Remission was defined as postoperative cortisol nadir of less than 2 mcg/dL and/or clinical/biochemical remission during follow-up.
Informed consent was obtained from parents and assent from patients if developmentally appropriate. Study procedures were approved by the NICHD and/or central National Institutes of Health Institutional Review Board.
Magnetic Resonance Imaging Scans
SPH was defined as minimal or no clinical symptoms reported by the patients (apart from those commonly associated with hypercortisolemia) and magnetic resonance imaging (MRI) findings consistent with hemorrhage, intratumor infarction, and/or intratumor cyst formation (suggesting old infarcts) based on the radiologist and the principal investigator’s (C.A.S.) assessment [12, 13]. MRI scans were performed based on standard clinical protocols as previously described [14]. Briefly, MRIs at the National Institutes of Health were performed before and after intravenous administration of gadolinium contrast material, with a gradient echo sequence and thin slices (≤ 1.5 mm). MRIs were performed in either a 1.5 Tesla or 3 Tesla MR machine from various manufacturers over time.
Clinical and Biochemical Data
Clinical and biochemical data were extracted from electronic medical records. Tumor size was recorded as the maximum dimension retrieved from the histology report. In cases where histologic report was not available, failed to identify a PA in the studied material, or if the histology report recorded only dimensions on fragments of the tumor, the maximum dimension was retrieved from the MRI images. If the MRI was negative and the histology was negative (but the patient achieved remission after surgery), the tumor size was recorded as missing.
Serum cortisol and plasma ACTH levels were calculated as the average value of the corresponding levels performed at 23:30 h and 00:00 h (reported as midnight values) and 07:30 h and 08:00 h (reported as morning values). Twenty-four–hour UFC was calculated as the average of the first 2 or 3 samples reported in the electronic medical records. Given the possible differences in the assays and reported reference range for UFC, we calculated the increase of UFC based on the upper limit of normal according to the following formula: UFC fold change = UFC/upper limit of the reference range. Serum cortisol was measured with solid-phase, competitive chemiluminescent enzyme immunoassay on a Siemens Immulite 2500 analyzer. Plasma ACTH was measured with a chemiluminescence immunoassay on a Siemens Immulite 200 XPi analyzer. UFC was measured with a chemiluminescent enzyme immunoassay until 2011 and with high-performance liquid chromatography–tandem mass spectrometry since 2011. High-dose dexamethasone suppression test was performed as previously described. Briefly, oral dexamethasone (120 mcg/kg, max 8 mg) was administered at 23:00 and cortisol was measured before (8 AM the day of administration) and after (9 AM the day after dexamethasone administration). The change of cortisol was calculated as: 100* [(postdexamethasone cortisol – predexamethasone cortisol)/predexamethasone cortisol]. Levels of cortisol lower than the lower limit of detection of the assay (< 1 mcg/dL) were substituted with the intermediate value (0.5) for all analyses. CRH stimulation test was performed as previously described. Briefly, an intravenous catheter was placed in the forearm the night before testing; the patient was fasting and lying in bed, and ovine CRH (oCRH) was administered (1 mcg/kg, max 100 mcg). Samples for cortisol and ACTH were taken at –5, 0, 15, 30, and 45 minutes after the administration of oCRH. The response to the last was expressed as the percentage change from baseline by subtracting the pretest cortisol and ACTH values from the posttest values and dividing by the former. The mean cortisol increase was estimated at 30 and 45 minutes from baseline. For ACTH the mean increase was estimated at 15 and 30 minutes after the administration of oCRH. CRH stimulation test was not performed after the discontinuation of oCRH by the company in November 2020.
Statistical Analysis
Categorical data are described as counts (proportions) and were compared between groups using the Fisher exact test. Fisher odds ratio (OR) was used to assess the odds of remission based on the presence of SPH and is presented as OR and 95% CI.
Continuous data were checked for normality and not normally distributed data are presented as median (first quartile to third quartile) and were compared between 2 groups using Wilcoxon rank-sum test. The Cox proportional hazards test was used to assess the risk of recurrence based on the presence of SPH and is presented as hazard ratio (HR) and 95% CI. Statistical analyses were performed in R.
Results
Clinical Data
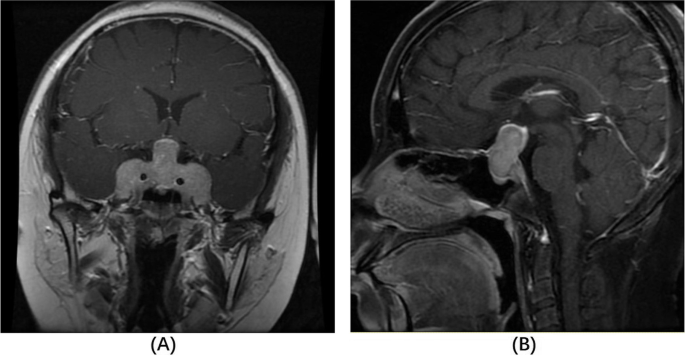
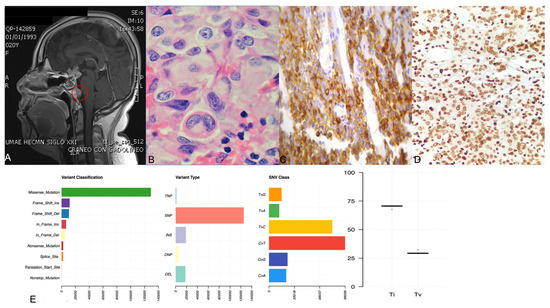
Out of 170 patients with available MRI before first surgery, 12 patients had evidence of possible SPH (7.1%) (Table 1). Various MRI findings were noted (Fig. 1), most commonly hyperintense lesions in T1 and T2 precontrast images (Fig. 1A and 1B), while some patients had intratumor heterogeneity suggestive of cystic formation (Fig. 1C-1F). As expected, the tumor size of patients with SPH as noted in histology reports or MRI images was higher than that in patients without SPH (median size: 8.5 mm [7.0-11.25] in the SPH group vs 5.4 mm [4-8] in the non-SPH group; P < .001).
Characteristics of patients with and without subclinical pituitary hemorrhage, necrosis, and/or cystic degeneration
| No SPH (N = 158) | SPH (N = 12) | P | |
|---|---|---|---|
| Age at diagnosis, y | 13.0 (10.6 to 15.4) | 12.5 (10.6 to 15.6) | .95 |
| Sex | |||
| Female | 89 (56.3%) | 5 (41.7%) | .49 |
| Male | 69 (43.7%) | 7 (58.3%) | |
| Disease duration, y | 2.50 (1.5 to 3.0) n = 138 |
1.00 (1.0 to 2.0) n = 10 |
.014 |
| Morning cortisol, mcg/dL | 16.2 (12.6 to 20.4) n = 139 |
18.4 (13.7 to 27.5) n = 10 |
.28 |
| Midnight cortisol, mcg/dL | 14.0 (10.7 to 19.7) n = 133 |
16.3 (11.0 to 23.0) n = 9 |
.52 |
| UFC fold change | 4.89 (2.51 to 8.50) n = 131 |
8.81 (6.86 to 9.42) n = 9 |
.18 |
| Morning ACTH, pg/mL | 39.4 (28.2 to 53.2) n = 143 |
60.8 (43.5 to 80.3) n = 10 |
.016 |
| Cortisol change during CRH stimulation test, % | 68.7 (33.3 to 111) n = 128 |
46.0 (–7.46 to 90.7) n = 8 |
.22 |
| ACTH change during CRH stimulation test, % | 145 (59.6 to 260) n = 127 |
103 (–5.75 to 278) n = 8 |
.55 |
| Cortisol change during high-dose dexamethasone suppression test, % | –85.8 (–90.5 to –76.8) n = 120 |
–58.0 (–85.4 to –49.7) n = 9 |
.035 |
| Remission | |||
| Yes | 127 (80.4%) | 10 (83.3%) | .99 |
| No | 25 (15.8%) | 2 (16.7%) |
Number in each cell reports the number of patients with available results.
Abbreviations: ACTH, adrenocorticotropin; CRH, corticotropin-releasing hormone; SPH, subclinical pituitary hemorrhage, necrosis, and/or cystic degeneration; UFC, urinary free cortisol.
Imaging findings in patients with subclinical pituitary hemorrhage, necrosis, and/or cystic degeneration. A, T1, and B, T2 magnetic resonance imaging (MRI) scans of the same patient showing area of high intensity inside the tumor suggesting acute/subacute episode. C, T2, and D, T1 MRI scans of the same patient showing heterogeneity in a large tumor with high-intensity areas suggesting blood-filled cavities and/or necrosis. E, T1, and F, T2 MRI scans of the same patient showing fluid inside the tumor.

Patients with and without SPH were similar in age (median age: 12.5 years [10.6-15.6] in the SPH group vs 13.0 years [10.6-15.4] in the non-SPH group; P = .95) and sex distribution (n of female = 5 (41.7%) in the SPH group vs 89 (56.3%) in the non-SPH group; P = .49). Patients with SPH had a shorter duration of disease as noted by changes in their growth chart parameters (median duration: 1.0 [1.0-2.0] year in the SPH group vs 2.5 [1.5-3.0] years in the non-SPH group; P = .014).
Patients in the 2 groups did not differ on their anthropometric characteristics, including height and body mass index z scores(P > .05). They also had similar blood pressure parameters and did not differ in terms of the frequency of hypertension diagnosis. No patient was on anticoagulation treatment nor had received radiation at the time of the MRI. One patient in the SPH group had a history of lower leg deep vein thrombosis and was previously on low-heparin therapy, but he had stopped treatment at least 6 months before the MRI.
Biochemical Evaluation of Hypercortisolemia
Morning and midnight serum cortisol and 24-hour UFC levels were similar in both groups, but patients with SPH had higher levels of morning ACTH (60.8 pg/mL [43.5-80.3]) compared to patients without SPH (39.4 pg/mL [28.2-53.2]; P = .016). Changes in cortisol and ACTH levels during the CRH stimulation test were similar between the 2 groups, but patients with SPH who underwent the overnight high-dose dexamethasone suppression test (n = 8) had lower suppression of cortisol after dexamethasone (–58.0% [–85.4 to –49.7]) compared to patients without SPH (n = 120) (–86.0% [–90.5 to –76.7]; P = .035) (Fig. 2). When the cutoff of suppression of more than 69% was considered, patients with SPH had a lower chance of suppressing more than 69% compared to patients without SPH (OR: 0.18; 95% CI, 0.03-0.95).
Cortisol levels before and after high-dose dexamethasone suppression test in patients with and without subclinical pituitary hemorrhage, necrosis, and/or cystic degeneration (SPH).

Remission After Surgical Treatment and Risk for Recurrence
The chance of immediate postoperative remission after surgery was similar in patients with and without SPH. For patients with initial remission and follow-up of at least 3 months, analysis of the risk of recurrence did not show a difference in recurrence rate between the 2 groups (HR: 1.12; 95% CI, 0.13-9.4, in the SPH group compared to the non-SPH group, adjusting for the neurosurgeon).
Discussion
SPH is often an incidental finding in the imaging evaluation of patients with PAs. The frequency of SPH in patients with CD is low (7.1% in our study) but these patients may differ in terms of their history of shorter duration of symptoms and the biochemical evaluation. More specifically, patients with CD and SPH showed higher ACTH levels and lower suppression of cortisol to high-dose dexamethasone. This, however, did not affect their prognosis in terms of immediate postoperative remission and long-term risk of recurrence.
SPH has been mainly studied in cohorts of patients with various types of PAs. From these studies important conclusions have been made suggesting that the risk of SPH is higher in patients with large nonfunctioning PAs or prolactinomas. Other risk factors for pituitary apoplexy are thought to be size of the adenoma, change in size, initiation, and withdrawal of dopamine agonists, type of dopamine agonist, use of anticoagulants, diabetes mellitus, hypertension, head trauma, radiotherapy, and preceding dynamic endocrine testing.
Patients with CD often represent a small portion of these cohorts, and to our knowledge there is no study to investigate how these patients respond to stimulation/suppression tests. For that reason, we evaluated these findings in our cohort of only patients with ACTH-secreting adenomas. As most of our referrals involve pediatric patients, we limited our cohort only to patients diagnosed at younger than 21 years to have a more homogeneous group.
The pathogenesis of pituitary apoplexy has been hypothesized to lie in more friable vessels in PAs, which, while the tumor increases in size, are more susceptible to rupture or cause surrounding feeding vessels to extend and bleed [7, 15]. CS, because of the coexisting hypercortisolemia, leads independently to endothelial dysfunction and coagulation defects, which are often apparent as easy bruising, thrombotic events, and other signs. However, review of the literature and the estimated frequency of SPH in our cohort suggest that patients with CD are not at increased risk for SPH, potentially related to the relatively small adenomas present in these patients [16, 17].
The main difference of patients with CD and SPH compared to those without lies in their biochemical testing, more characteristically in lower suppression to dexamethasone. The overnight high-dose dexamethasone suppression test was originally designed to differentiate various types of CS [18]. Although originally described as a highly accurate test, in clinical practice, cutoffs of 50% to 80% have shown variable sensitivity and specificity and certain centers opt not to use this test unless all other diagnostic evaluations yield confounding results [19-21]. In previous studies a threshold of suppression of more than 69% showed the highest accuracy (sensitivity: 71%, specificity: 100%), and we have incorporated this in our diagnostic algorithm (acknowledging the limitations of the test) [22, 23]. In our analysis, patients with SPH had lower suppression of cortisol under the effect of high-dose dexamethasone and a higher chance of not passing the aforementioned threshold. The mechanism for the lower suppression to dexamethasone of these tumors may be due to lower vascular circulation of dexamethasone at the level of the tumor, and/or the lower sensitivity of necrotic cells to the negative feedback by circulating glucocorticoids.
A limitation of this study was that the diagnosis of SPH was based on MRI findings. However, MRI sequences and machines differed between patients and over time. Thus, although large hemorrhagic/necrotic lesions are probably accurately identified, it is possible that smaller lesions are misclassified as negative; the effect of smaller hemorrhagic areas to the biochemical testing may however be smaller as well. Further, the MRIs were not read by a central radiologist, but rather from the radiologist on call at each time point, and this could lead to discrepancies in readings. In addition, our cohort’s data may not be generalized to the pediatric or adult CD population, as often our referrals consist of patients with difficult to treat, small, or otherwise unusual tumors.
In conclusion, SPH may be incidentally identified in up to 7% of patients with CD. Patients with CD and SPH may differ in terms of their response to endocrine tests, and this finding should be incorporated in their evaluation.
Abbreviations
-
ACTH
adrenocorticotropin
-
CD
Cushing disease
-
CRH
corticotropin-releasing hormone
-
CS
Cushing syndrome
-
HR
hazard ratio
-
MRI
magnetic resonance imaging
-
NICHD
Eunice Kennedy Shriver National Institute of Child Health and Human Development
-
OR
odds ratio
-
PA
pituitary adenoma
-
SPH
subclinical pituitary hemorrhage
-
UFC
urinary free cortisol
Financial Support
This work was supported by the intramural research program of the Eunice Kennedy Shriver NICHD, NIH, Bethesda, MD 20892, USA.
Disclosures
Dr Stratakis holds patents on the function of the PRKAR1A, PDE11A, and GPR101 genes and related issues; his laboratory has also received research funding on the GPR101 gene, and on abnormal growth hormone secretion and its treatment by Pfizer, Inc. He is currently employed by ELPEN, SA and has been consulting for Lundbeck Pharmaceuticals and Sync, SA. The other authors have nothing to disclose.
Data Availability
Some or all datasets generated during and/or analyzed during the current study are not publicly available but may be available from the corresponding author on reasonable request.
Filed under: Cushing's, pituitary | Tagged: ACTH, hemorrhage, pituitary | Leave a comment »


{kind=link}
{kind=link}