Endogenous hypercortisolism (Cushing syndrome) is a multisystemic disease characterized by a wide range of clinical signs and symptoms. Its heterogeneous presentation can cause significant diagnostic delays, and prolonged exposure to excess cortisol activity can contribute to cardiometabolic abnormalities such as diabetes. When diabetes remains unresponsive or only partially responsive to standard-of-care treatment, clinicians should consider hypercortisolism as a potential underlying driver.Despite the risks associated with hypercortisolism, guidance on identifying and managing it in patients with diabetes remains limited. This article presents a case series of 10 patients from a single practice who were screened for hypercortisolism because of difficult-to-manage diabetes and additional comorbidities. All patients were treated for hypercortisolism with mifepristone, resulting in significant clinical improvements including weight loss, improved glycemic control, and reduced medication needs.
This real-world case series highlights the importance of recognizing hypercortisolism as a differential diagnosis and a potential contributing factor to difficult-to-manage diabetes despite standard-of-care therapies. Addressing hypercortisolism with mifepristone can result in substantial clinical benefits.
Double Cushing syndrome exists: exogenous steroid use can mask concurrent adrenal hypercortisolism. When symptoms persist and cortisol remains high after tapering or stopping prescribed glucocorticoids, an endogenous source is likely. Early recognition with ACTH testing, dexamethasone suppression, and adrenal imaging reduces misdiagnosis, favors timely surgery, and supports safe tapering.
1 Introduction
Cushing syndrome (CS) is a non-physiological increase in plasma glucocorticoids [1]. In most cases, the source of increased plasma glucocorticoids is caused by exogenous steroid administration, which is quite common, and about 1% of the world population is on long-term (more than 3 months) oral glucocorticoids [1, 2]. On the contrary, endogenous overproduction of glucocorticoids is rare, and annually, only two to eight per million people are diagnosed with endogenous CS [3]. The simultaneous occurrence of endogenous and exogenous CS is an exceptionally uncommon phenomenon. This dual manifestation has been reported in a few case reports, highlighting its rarity and the complex diagnostic and therapeutic challenges it poses [4, 5]. Therefore, in this study, we discuss a patient who presented with cushingoid features and was simultaneously diagnosed with both endogenous and exogenous CS or, as it is called, double CS.
2 Case Presentation
The patient was a 46-year-old male with a history of new-onset hypertension and recurrent deep vein thrombosis (DVT) who was referred to our endocrinology clinic with a chief complaint of hip pain and weakness of the lower limbs. In the past 3 years, the patient had been receiving 50 mg/day of oral prednisolone and inhalation powder of Umeclidinium and Vilanterol (62.5/25 μg/dose) because of respiratory complications that started after Coronavirus Disease 2019 (COVID-19) vaccination. After 3 months of corticosteroid treatment, he experienced DVT for the first time when he was started on rivaroxaban. However, while he was on treatment, the second DVT occurred 1 month before his referral, and therefore, rivaroxaban was changed to warfarin 5 mg/day.
The patient also mentioned weight gain with his body mass index (BMI) rising from 26 to 31 kg/m2, progressive weakness of proximal muscles, easy bruising, decreased libido, mood changes with mostly euphoric mood, and irritability during the last 2 years. Moreover, multiple osteoporotic fractures of ribs, clavicle, sternum, and lumbar vertebrae were added to his symptoms in the past 5 months. At that time, he underwent bone densitometry, which revealed osteopenia of the left hip with a Z-score of −1.3 and severe osteoporosis of total lumbar spine with a T-score of −3.9. He started taking calcium and vitamin D3 supplements and received a single injection of 750 μg/3 mL teriparatide 30 days before his referral to our center.
Two months ago, the patient gradually reduced the dosage of prednisolone by tapering the dose to 12.5 mg/day. However, a month later, the hip pain and muscle weakness worsened to such an extent that the patient was unable to walk. Due to his signs and symptoms, the patient was referred to our center for further evaluation of CS. The patient also mentioned a history of nephrolithiasis, new-onset hypertension, and lower limb edema, for which he was started on eplerenone 25 mg and furosemide 20 mg tablets once daily. In his family history, the patient’s mother had type 2 diabetes mellitus, and his two sisters had a history of nephrolithiasis. The patient did not mention any history of allergies to medications or foods. He was addicted to opium and had 15 pack-years of smoking, but he did not mention alcohol consumption.
Upon admission, the patient presented with a blood pressure of 150/83 mmHg, heart rate of 74 bpm, respiratory rate of 20/min, temperature of 36.5°C, oxygen saturation of 93%, and BMI of 31 kg/m2. He was sitting in a wheelchair due to weakness and severe pain in the hip. On physical examination, the patient showed the features of CS, including moon face, buffalo hump, central obesity, facial plethora, thin and brittle skin, acne, and purple stretch marks (striae) on the flanks (Figure 1). Proximal muscle weakness in the lower limbs with a muscle force grade of 4/5 and 3+ edema was also observed. Laboratory investigations are shown in Table 1.
De-identified clinical photographs illustrating the Cushingoid phenotype. (A) Overall habitus with marked central (truncal) adiposity. (B) Rounded plethoric face (“moon facies”). (C) Relatively slender distal extremities compared with truncal obesity. (D) Dorsocervical fat pad (“buffalo hump”). (E) Upper thoracic/supraclavicular fat accumulation. (F) Protuberant abdomen with wide violaceous striae.
TABLE 1. Laboratory findings of case report.
Laboratory test
Patient value (in-hospital)
Patient value (follow-up)
Reference range
On admission
Hemoglobin (g/dL)
16.6
13.6
13.5–17.5
Hematocrit (%)
49.5
42.1
42–52
WBC (white blood cells; 103/μL)
11.8
7.1
4.0–11.0
PLT (platelet count; 103/μL)
286
294
150–450
BUN (blood urea nitrogen; mg/dL)
10
11
7–18
Cr (creatinine; mg/dL)
0.9
0.9
0.7–1.3
ALP (alkaline phosphatase; IU/L)
1016
129
44–147
AST (aspartate aminotransferase; IU/L)
48
30
< 31
ALT (alanine transaminase; IU/L)
88
21
< 31
CRP (C-reactive protein; mg/dL)
31
3
< 5
ESR (erythrocyte sedimentation rate; mm/h)
63
24
< 15
Sodium (mEq/L)
148
141
136–145
Potassium (mEq/L)
4.8
4.3
3.5–5
FBS (fasting blood glucose; mg/dL)
97
89
80–100
TC (total cholesterol; mg/dL)
267
182
< 200
TG (triglyceride; mg/dL)
148
104
< 200
LDL (low-density lipoprotein; mg/dL)
138
98
< 130
HDL (high-density lipoprotein; mg/dL)
64
55
30–70
In hospital
Cortisol 8 a.m. fasting (μg/dL)
20.2
14.1
4.3–24.9
ACTH (adrenocorticotropic hormone; pg/mL)
< 1
—
7.2–63.3
1 mg Overnight dexamethasone suppression test (μg/dL)
16.5
—
< 1.8
3 Methods (Differential Diagnosis, Investigations, and Treatment)
Initially suspected of having exogenous-induced CS, the patient’s prednisolone was on hold for 3 days. Cortisol 8 a.m. fasting level, measured with Electrochemiluminescence (ECL) and adrenocorticotropic hormone (ACTH) test, was 20.2 μg/dL (585.4 nmol/L) and < 1 pg/mL, respectively. Due to the lack of suppression of serum cortisol despite not using oral glucocorticoids, the absence of adrenal insufficiency symptoms, and the fact that the patient’s symptoms remained unchanged during this period, co-occurrence of endogenous CS was suspected.
A 1 mg overnight dexamethasone suppression test was performed to confirm endogenous CS diagnosis, and the results were reported as 16.5 μg/dL (normal range < 1.8 μg/dL). Considering the possibility of an ACTH-independent CS, the patient underwent an abdominopelvic multidetector computed tomography (MDCT) of abdominopelvic with adrenal protocol, which revealed a well-defined lesion with an approximate size of 32.8 × 38.6 mm in the left adrenal gland with a radiodensity of 90 Hounsfield units and a normal right adrenal gland (Figure 2). Moreover, evidence of previous old fractures as multiple callus formation was seen involving the clavicles, sternum, bilateral ribs, ischium, and pelvic bones. Multilevel old stable compression fractures of thoracic and lumbar vertebral bodies were also present. The differential diagnoses were glucocorticoid secretory adrenal tumors, including adrenal cell carcinoma and lipid-poor adenoma. In order to rule out pheochromocytoma, 24-h urine catecholamines were measured, and the results were negative.
Abdominopelvic multidetector computed tomography (MDCT) with adrenal protocol showing a well-defined lesion with an approximate size of 32.8 × 38.6 mm in the left adrenal gland; radiodensity 90 HU. (A) Transverse plane. (B) Coronal plane. (C) Sagittal plane.
Finally, the patient underwent left adrenalectomy and corticosteroid replacement therapy due to the suppression of the other adrenal gland. According to the post-operative pathological investigations, immunohistochemistry markers reported as negative chromogranin, positive melan-A and inhibin, less than 3% Ki-67 marker, and lipid-poor adrenal cortical adenoma without invasions were diagnosed (Figure 3).
Immunohistochemistry of the adrenal lesion (all panels acquired with a 100× oil-immersion objective; 10× eyepiece; original magnification ×1000). (A) Positive inhibin, (B) Positive Melan-A, (C) Less than 3% Ki-67 marker, and (D) Negative chromogranin.
4 Results (Outcome and Follow-Up)
Within 3 months after the operation, the patient’s corticosteroid was tapered and then discontinued due to the normalization of the cortisone serum test (14.1 μg/dL). Proximal limb weakness and hip pain, which had deprived the patient of the ability to move, gradually improved so that he could walk easily and perform daily activities. The signs and symptoms related to CS, including the patient’s mood, skin signs, and general appearance, returned to normal. The patient has been followed up for 6 months after the surgery. The patient’s BMI decreased to 24 kg/m2, and he stopped his anti-hypertensive medications with a blood pressure of 100/60 mmHg without previously prescribed drugs. So far, the laboratory tests have been within the normal range, and he has no complaints (Table 1).
5 Discussion
The described case was diagnosed with a cortisol-producing adrenocortical adenoma accompanied by exogenous CS. CS is an uncommon clinical condition caused by prolonged exposure to increased cortisol levels, which can be due to endogenous or exogenous factors [6]. Endogenous CS is infrequent and is classified as ACTH-dependent (80% of cases) or ACTH-independent (20% of cases) [7]. In the ACTH-independent category, adrenal adenoma accounts for 60% of cases and only 12% of cases of endogenous CS [7, 8]. Exogenous CS mainly occurs due to prolonged administration of glucocorticoids, which are used to manage a broad spectrum of diseases such as inflammatory, autoimmune, or neoplastic disorders and are the most common cause of CS worldwide [9]. Multiple factors, including formulation, duration of administration, pharmacokinetics, affinity, and potency of exogenous glucocorticoids, affect the probability of exogenous CS, but all forms of glucocorticoids can induce CS [10].
In the setting of cushingoid clinical features with chronic administration of high-dose glucocorticoids, especially oral prednisolone, the probability of exogenous CS is remarkably high; therefore, CS diagnostic approaches suggest that the first step after confirmation of cortisol excess is ruling out exogenous glucocorticoid administration [7, 8, 10]. Therefore, the possibility of co-occurrence of endogenous CS with iatrogenic CS is extremely low, and the diagnosis requires high clinical suspicion [4].
Differentiating endogenous and exogenous CS based on clinical features can be challenging and far-fetched. However, a few points can help physicians distinguish between these two. First, exogenous CS symptoms tend to be more striking, while endogenous CS appears more gradually. Second, hypertension, hypokalemia, and features of androgen excess, such as acne and hirsutism, are more common in endogenous CS [4, 10]. In addition, endogenous CS should be suspected if the patient’s symptoms continue after corticosteroid discontinuation or if the serum cortisol level is high despite corticosteroid cessation. In our case, the patient had a high cortisol level despite stopping prednisolone for 3 days, and he did not have any symptoms of adrenal insufficiency despite stopping prednisolone suddenly. Consequently, it was suspected that glucocorticoids might come from an endogenous source. Because ACTH was suppressed concurrently with elevated cortisol, non-ACTH-dependent CS was suspected, and MDCT of abdominopelvic confirmed it.
So far, few similar cases of simultaneous endogenous and exogenous CS have been reported. The first case was a 23-year-old woman with juvenile idiopathic arthritis who was administered high doses of triamcinolone for 16 years [4]. The development of cushingoid features that favored endogenous CS, such as hirsutism and acne, strengthened the suspicion of endogenous CS, and a CT scan revealed hypercortisolism with a bulky and nodular left adrenal gland, and a double CS was confirmed [4]. The second case was a 66-year-old woman diagnosed with exogenous CS after consumption of Traditional Chinese medicine (TCM) for a year [5]. The cessation of TCM did not significantly improve her cushingoid features, and she developed additional CS complications, including hypertension, diabetes mellitus, and osteoporotic fractures over the next 8 years. CS workup revealed a right-sided adrenal adenoma, and after the adrenalectomy, her clinical cushingoid features markedly improved [5]. These cases suggest that exogenous and endogenous CS can exist simultaneously in the same person. Although it is very rare, it should be considered in a person who still complains of CS symptoms after corticosteroid cessation. We suggest clinicians evaluate the patients for the disappearance of exogenous CS symptoms after tapering and stopping glucocorticoids. If the symptoms remain, they should be evaluated for endogenous CS.
6 Conclusion
The co-occurrence of an endogenous CS in the setting of an exogenous CS is curious. The diagnosis is based on a high clinical suspicion. Clinicians should evaluate patients for symptom resolution after tapering and discontinuing corticosteroids. Clinical cushingoid features that do not resolve after discontinuing exogenous glucocorticoids and high cortisol levels despite discontinuing corticosteroids should raise clinicians’ suspicion of the co-occurrence of exogenous and endogenous CS.
Author Contributions
Reza Amani-Beni: investigation, methodology, writing – original draft, writing – review and editing. Atiyeh Karimi Shervedani: methodology, writing – original draft. Bahar Darouei: conceptualization, validation, writing – review and editing. Matin Noroozi: methodology, writing – original draft. Maryam Heidarpour: conceptualization, supervision, validation, writing – review and editing.
Acknowledgments
The authors have nothing to report.
Consent
Written informed consent was obtained from the patient to publish this report, including de-identified clinical photographs, in accordance with the journal’s patient consent policy.
Conflicts of Interest
The authors declare no conflicts of interest.
Data Availability Statement
The data that supports the findings of this study are available on request of the corresponding author. The data are not publicly available due to privacy restrictions.
Osilodrostat’s clinical development program mostly enrolled Cushing disease patients. Data in non-pituitary Cushing syndrome (CS) are limited.
Objective
Evaluate osilodrostat effectiveness and safety in non-pituitary CS in real-world practice in France.
Design
Retrospective, observational study (LINC 7; NCT05633953). Data for patients who initiated osilodrostat under the French Autorisation Temporaire d’Utilisation scheme or, once approved, in routine clinical practice were extracted retrospectively for ≤36 months (2019–2022).
Setting
Multicenter institutional practice.
Patients or Other Participants
103 adult non-pituitary CS patients: ectopic adrenocorticotropic hormone secretion (EAS), n=53; adrenocortical carcinoma (ACC), n=19; adrenal adenoma (AA), n=17; bilateral adrenal nodular disease (BND), n=14. 43 remained on osilodrostat throughout the observation period.
Intervention
Median (min–max) osilodrostat exposure and baseline dose: 177 days (1–1178), 5.0 mg/day (1–60).
Main Outcome Measure
Proportion with mean urinary free cortisol (mUFC) ≤ upper limit of normal (ULN) at week 12 (modified intention-to-treat [mITT] population: all enrolled patients with ≥12W follow-up, excluding patients without W12 mUFC for non-safety reasons).
Results
Osilodrostat was initiated and titrated based on investigator judgment. Cortisol decreased by W4, remaining stable thereafter. 23/52 patients (mITT; 44.2%, 95% CI 30.5–58.7) had mUFC ≤ULN at W12 (missing values input as non-responders). 45/52 had W12 mUFC available; proportion with mUFC ≤ULN by etiology: EAS, n=12/29 (41%); ACC, n=4/6; AA, n=1/3; BND, n=6/7. Most common (≥15%) TEAEs: adrenal insufficiency (28%) and hypokalemia (18%). 29 patients (EAS, n=24; ACC, n=5) died from AEs (n=1 assessed as osilodrostat related by investigator), mostly neoplasm progression (n=11).
Conclusions
Osilodrostat is a suitable treatment for endogenous Cushing syndrome of various non-pituitary etiologies.
Accepted manuscripts are PDF versions of the author’s final manuscript, as accepted for publication by the journal but prior to copyediting or typesetting. They can be cited using the author(s), article title, journal title, year of online publication, and DOI. They will be replaced by the final typeset articles, which may therefore contain changes. The DOI will remain the same throughout.
Cushing syndrome (CS) impairs quality of life (QoL) and mood. Prospective real-life data on post-treatment recovery and predictors of improvement are limited.
Objectives
Evaluate changes in QoL, depression, and anxiety in patients with CS, before and after biochemical control, and identify predictors of clinically meaningful improvement.
Design and Setting
Prospective observational study at a tertiary center.
Patients
67 patients with endogenous CS (60 pituitary, 7 adrenal) were assessed with active disease and again after achieving biochemical control through surgery and/or medication.
Outcomes
Patient-reported outcomes included CushingQoL, Beck Depression Inventory-II (BDI-II), and State-Trait Anxiety Inventory (STAI).
Results
Mean and longest follow-up was 2.3 and 11.5 years, respectively. Treatment led to improvements in mean scores across all domains (QoL: +18.2±20.9, BDI: –6.8±8.6, STAI-State: –9.6±12.5, STAI-Trait: –8.6±12.6; all p < 0.001). However, minimal important difference was achieved in 64.6% for QoL, 67.9% for BDI, 53.2% and 52.8% for STAI subscales. After multivariable analysis, QoL improvements were predicted by lower baseline BMI, pre-treatment symptoms ❤ years, post-operative hydrocortisone replacement >6 months, and normal follow-up late-night salivary cortisol (LNSC). Depression improvements were predicted by symptoms ❤ years, normal follow-up LNSC, and surgical treatment. Anxiety improvements were predicted by younger age and >6 months post-operative hydrocortisone. Depression improved more gradually than QoL and anxiety.
Conclusions
Although effective treatment improves mood and QoL in CS, clinically meaningful recovery is variable and incomplete for some patients. Our findings highlight the need to limit diagnostic delay and provide comprehensive post-treatment care that includes normalization of cortisol circadian rhythm.
Accepted manuscripts are PDF versions of the author’s final manuscript, as accepted for publication by the journal but prior to copyediting or typesetting. They can be cited using the author(s), article title, journal title, year of online publication, and DOI. They will be replaced by the final typeset articles, which may therefore contain changes. The DOI will remain the same throughout.
This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivs licence (https://creativecommons.org/licenses/by-nc-nd/4.0/), which permits non-commercial reproduction and distribution of the work, in any medium, provided the original work is not altered or transformed in any way, and that the work is properly cited. For commercial re-use, please contact reprints@oup.com for reprints and translation rights for reprints. All other permissions can be obtained through our RightsLink service via the Permissions link on the article page on our site—for further information please contact journals.permissions@oup.com. See the journal About page for additional terms.
IntroductionCushing’s disease (CD) is the most common cause of endogenous Cushing’s syndrome. Adrenocorticotropic hormone (ACTH) has trophic and mitogenic effects on the adrenal cortex that may cause diffuse adrenal enlargement and nodular lesions.
AimTo evaluate the prevalence of adrenal structural abnormalities in patients with CD.
MethodsRetrospective cohort study. We conducted a computerized search in our medical centers databases for the diagnosis of CD recorded between the years 1995–2024. Out of 124 patients with ACTH dependent Cushing’s syndrome, we identified 68 patients with CD who underwent adrenal imaging. We analyzed the clinical, biochemical, and imaging data.
ResultsOur cohort included 68 patients (51 females, 75.0%; mean age at the time of adrenal imaging, 44.6 ± 14.9 years). Sixteen (23.5%) patients had an adrenal nodule ≥10 mm (median size, 27.5 mm, IQR 14.3–38.3), and 19 others (27.9%) had adrenal hyperplasia or nodules <10 mm. The prevalence of adrenal nodules increased with age from 16.7% in patients aged 26–35 years to 26.3% in those aged above 55. Patients with adrenal nodules were older compared to those with normal adrenal glands (mean age, 49.0 ± 12.4 vs 39.1 ± 14.9 years; p = 0.03), and had lower ACTH level (0.7 x ULN, IQR 0.5–1.2, vs 1.2 x ULN, IQR 0.9–1.8, p = 0.02).
ConclusionsWe identified abnormal adrenal imaging in 51.5% of patients with CD. The prevalence of adrenal nodules in our study was 10-fold higher than in the normal population, for all age groups. This suggests that chronic ACTH secretion in CD is associated with adrenal nodules appearance.
Explore related subjects
Discover the latest articles, books and news in related subjects, suggested using machine learning.
Cushing’s disease (CD) is the most common etiology (70%) of endogenous Cushing’s syndrome [1]. CD is caused by a pituitary adenoma that autonomously secretes adrenocorticotropic hormone (ACTH), leading to cortisol overproduction and secretion from the adrenal cortex [2].
ACTH is a predominant trophic factor of the adrenal cortex. Several animal models, in which ACTH or its receptor (melanocortin 2 receptor, MC2R) were eliminated, have confirmed the central role of ACTH in maintaining normal growth of the adrenal cortex [3, 4]. While ACTH also exerts a mitogenic effect, the precise mechanism by which it promotes adrenocortical growth and proliferation is complex and only partially understood [5]. Due to the effects of ACTH on the adrenal cortex, patients with ACTH-dependent Cushing’s syndrome have high prevalence of adrenal hyperplasia, reaching up to 60% [6, 7].
Adrenal incidentaloma (AI) is an adrenal mass (≥1 cm) detected on imaging not performed for a suspected adrenal disease [8]. Autopsy studies suggest that the overall prevalence of adrenal masses ranges from 1.1–8.7%, which increases with age [9]. In recent decades, with advancement in imaging technologies, radiological studies have become more common and accurate, with the prevalence of AI’s in imaging studies approaching values similar to those found in autopsy studies [8, 10,11,12,13,14].
In contrast to adrenal hyperplasia, there are limited studies examining the prevalence of adrenal nodules in CD. These studies, which included small cohorts of up to 40 patients with CD, found a significantly higher prevalence of adrenal nodules, with rates ranging from 4% to nearly 40% among studied individuals [6, 7, 15].
Our aim was to study the morphology of the adrenal glands and assess the prevalence of abnormal adrenal findings, including hyperplasia and adrenal nodules, in a large cohort of patients with CD.
Materials and methods
We conducted a computerized search in Rabin and Shamir Medical Centers databases for the diagnosis of Cushing’s syndrome and screened for patients with ACTH-dependent Cushing’s syndrome. Cushing’s syndrome was diagnosed in patients with characteristic symptoms and signs and hypercortisolemia. Hypercortisolemia was confirmed according to laboratory findings including high 24 h urinary free cortisol (UFC), elevated midnight salivary cortisol, and abnormal 1 mg dexamethasone suppression test. We further classified these patients to ACTH-dependent Cushing’s syndrome, based on ACTH level in the normal or above the normal range.
A diagnosis of CD was confirmed by a pituitary adenoma of ≥6 mm depicted by seller MRI, an inferior petrosal sinus sampling (IPSS) supporting a pituitary source of ACTH secretion, immunostaining for ACTH and/or T-PIT in resected tumor specimens, and/or hormonal remission following successful trans-sphenoidal adenoma resection.
After identifying all patients with CD, our cohort included only patients with CD who have undergone abdominal computed tomography (CT) or abdominal magnetic resonance imaging (MRI), during the active phase of their disease.
In addition, we assembled a cohort of patients with ACTH-dependent Cushing’s syndrome in whom the source of ACTH secretion was not identified, that is, patients with ACTH-dependent Cushing’s syndrome without tumor localization.
Patients with malignant pituitary tumor and those with confirmed ectopic ACTH secretion were excluded. Patients that were treated with glucocorticoids were also excluded.
Based on an imaging report from an expert radiologist, all adrenal images were classified into three categories: normal, hyperplastic, or nodular adrenal glands. All adrenal nodules were classified according to nodule size: maximal nodule diameter below 10 mm, or ≥10 mm.
The study was approved by the Rabin Medical Center and Shamir Medical Center institutional review boards with waiver of patient consent, as complied with the Helsinki Declaration.
The authors received no funding for performing this study.
Statistical analysis and plan
Statistical analysis was performed using IBM SPSS version 29.0 (IBM Corp., Armonk, NY).
Continuous variables were presented by Mean (SD) or Median (IQR) as appropriate. Dichotomous variables were presented by N (%).
T-test and Mann–Whitney tests were used to compare values of normally and non-normally distributed continuous variables, with Chi-Square test used for comparison of categorical variables.
Two-sided P-values less than 0.05 were considered statistically significant.
Results
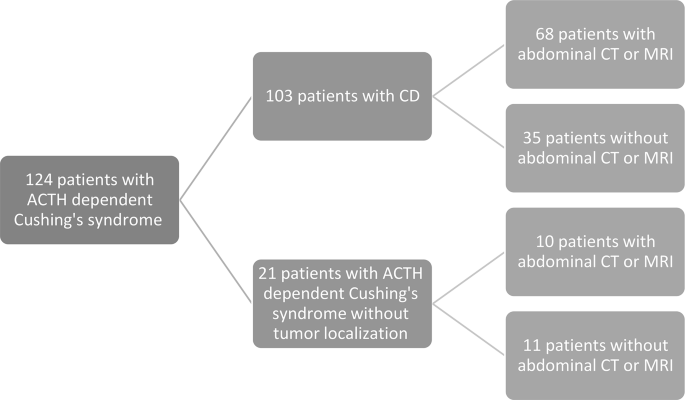
From March 1995 to December 2024, a total of 124 patients with ACTH-dependent Cushing’s syndrome were identified. There were 103 patients with the diagnosis of CD, and 21 patients with ACTH-dependent Cushing’s syndrome without tumor localization. After carefully reviewing each case, we excluded 35 patients without reported adrenal imaging (Fig. 1).
Fig. 1
Patient selection flowchart. ACTH adrenocorticotropic hormone, CD Cushing’s disease, CT computed tomography, MRI magnetic resonance imaging
The main cohort included 68 patients with CD and available adrenal imaging (51 females, 75.0%; mean age at the time of adrenal imaging, 44.6 ± 14.9 years). The median pituitary adenoma size (i.e. largest adenoma diameter) was 6 mm (IQR 4.75–10.25). The median ACTH level was 1.2 x ULN (IQR 0.8–1.9), and the median UFC level was 3.5 x ULN (IQR 2.0–6.0) (Table 1).
Table 1 Baseline characteristics of 68 patients with Cushing’s disease and 10 subjects with ACTH dependent Cushing’s syndrome without tumor localization
Sixty-five (95.6%) patients underwent trans-sphenoidal surgery (TSS), 44 of them (67.7%) had a resected adenoma expressing ACTH and/or T-PIT, 7 (10.8%) had no pituitary adenoma in pathology, and for 14 (21.5%) patients we did not have available pathology reports. Forty-six of the 65 (70.8%) patients experienced hormonal remission following surgery (Table 1).
During follow-up, 17 (26.2%) patients underwent repeated pituitary surgery, 8 of them due to persistent disease, and 9 due to recurrent elevated cortisol. Fifteen (22.1%) patients underwent radiation therapy. Thirty-nine (57.4%) patients received medical therapy for CD with adrenal steroidogenesis inhibitors and medications targeting pituitary somatostatin or dopamine receptors. Of these, seven (17.9%) patients were treated medically before TSS, 31 (79.5%) after TSS, and one (2.6%) patient received medical therapy both before and after TSS (Table 1).
Abdominal imaging findings in patients with CD
Sixty-three patients had abdominal CT, including five who had Ga68 positron emission tomography (PET) CT without pathologic adrenal Ga68 uptake on functional imaging. Five additional patients had abdominal MRI.
Twenty-three (33.8%) patients underwent adrenal imaging as part of their medical evaluation following the diagnosis of Cushing’s syndrome. Five (7.4%) patients underwent adrenal imaging due to persistent or recurrent disease following surgery. Eleven (16.2%) patients had adrenal imaging because of abdominal pain, thirteen (19.1%) patients for other reasons not related to Cushing’s syndrome (e.g. following abnormal finding in ultrasonography (US), or before abdominal/gynecological surgery) and sixteen patients (23.5%) for unknown reasons.
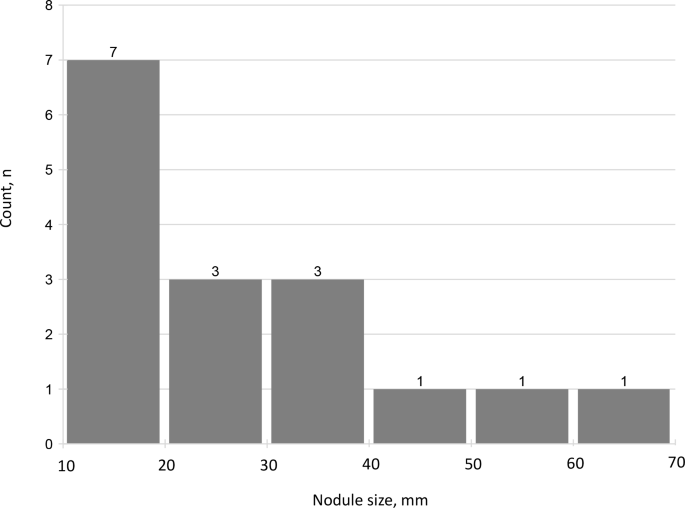
A total of 16 (23.5%) patients had an adrenal nodule ≥10 mm (median size, 27.5 mm, IQR 14.3–38.3) (Table 1). All of the nodules were radiologically defined as compatible with adenomas, based on low Hounsfield Units (HU) on non-contrast CT or signal drop on out-of-phase MRI sequences. Five patients had nodules in the right adrenal gland, six others had nodules in the left adrenal gland, and five patients had adrenal nodules ≥10 mm in both adrenal glands – one nodule in each side. There was only one patient with adrenal imaging consistent with bilateral multinodular adrenal hyperplasia, that was classified into the group of patients with adrenal nodule ≥10 mm. Nine (13.2%) patients had an adrenal nodule ≥20 mm (Fig. 2).
Nineteen (27.9%) patients had adrenal hyperplasia and/or nodules smaller than 10 mm (Table 1).
Patients with adrenal nodules ≥10 mm, as well as those with adrenal hyperplasia and/or nodules <10 mm, were significantly older compared to individuals with normal adrenal glands (mean age at imaging: 49.0 ± 12.4 and 50.4 ± 13.7 vs 39.1 ± 14.9 years, respectively; p = 0.03 and p = 0.01). Only patients with adrenal nodules ≥10 mm had significantly lower ACTH levels compared to patients with normal adrenal glands (0.7 x ULN, IQR 0.5–1.2, vs 1.2 x ULN, IQR 0.9–1.8; p = 0.02) (Table 2).
Table 2 Clinical characteristics of CD patients with adrenal nodules ≥10 mm, adrenal hyperplasia and/or nodule <10mm and with normal adrenal glands
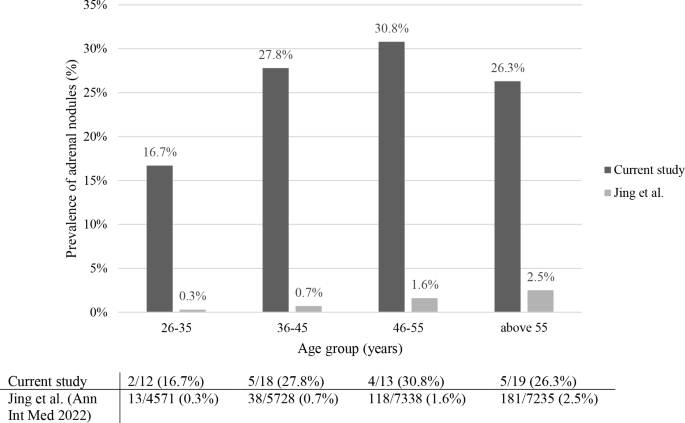
The prevalence of adrenal nodules increased with age from 16.7% in patients aged 26–35 years to 27.8, 30.8 and 26.3% in those aged 36–45, 46–55, and above 55 years, respectively (Fig. 3).
Fig. 3
Prevalence of adrenal nodules ≥10 mm in CD patients (current study) and in the general population (Jing et al.), stratified by age group
Urinary free cortisol (UFC) levels did not differ significantly among the three groups. Patients with adrenal nodules ≥10 mm had a median UFC level of 4.0 x ULN (IQR 1.7–6.3), those with adrenal hyperplasia and/or nodules <10 mm had a median of 3.8 x ULN (IQR 3.0–6.0), and patients with normal adrenal glands had a median of 3.5 x ULN (IQR 2.0–6.0) (p = 0.77 and p = 0.63, respectively vs. normal adrenal glands) (Table 2).
A pituitary adenoma was depicted by sellar MRI in 54 (79.4%) patients. Patients with adrenal nodules ≥10 mm tended to harbor a larger pituitary adenoma compared to patients with normal adrenals (median adenoma size, 7.0 mm (IQR 6.0–11.5) vs 6.0 mm (IQR 4.0–8.0) respectively; p = 0.11). Patients with adrenal hyperplasia and/or nodules <10 mm had a median adenoma size of 5.5 mm (IQR 3.0–13.0), which was not significantly different compared to patients with normal adrenal glands (p = 0.63) (Table 2).
Remission rates after TSS were not significantly different among the groups. Among patients with adrenal nodules ≥10 mm, 12 (80%) patients achieved remission, compared to 23 (71.9%) patients with normal adrenal glands (p = 0.73). Similarly, 11 patients (61.1%) with adrenal hyperplasia and/or nodules <10 mm achieved remission, with no significant difference compared to patients with normal adrenal glands (p = 0.53) (Table 2).
Clinical characteristics of four patients with CD who underwent unilateral adrenalectomy
Four patients (all females; mean age at the time of adrenal imaging, 46.3 ± 6.8 years) underwent unilateral adrenalectomy for a benign adrenocortical adenoma (adrenal adenoma size between 2.8–6.4 cm). One patient underwent adrenalectomy prior to TSS, with persistent hypercortisolism following adrenal surgery but achieved remission after TSS (Table 3).
Table 3 Clinical characteristics of four patients with CD who underwent unilateral adrenalectomy
Three patients underwent adrenalectomy after TSS. All three patients exhibited persistent hypercortisolism prior to adrenalectomy, with a median UFC level of 3.0 x ULN (IQR 1.9–6.1). The median ACTH level was 0.6 x ULN (IQR 0.3–1.1). One of them experienced transient postoperative cortisol normalization, while the other two achieved remission following adrenalectomy (Table 3).
Abdominal imaging findings in patients with ACTH-dependent Cushing’s syndrome without tumor localization
Ten patients (including 7 females) with ACTH-dependent Cushing’s syndrome without tumor localization had adrenal imaging. Compared to patients with CD, these patients were older (mean age at the time of adrenal imaging, 58.4 ± 15.1 vs 44.7 ± 14.9 years, p < 0.01). Three of these patients (30.0%) had adrenal nodule ≥10 mm (median size, 14 mm, IQR 11.7–18), and 6 (60.0%) had adrenal hyperplasia or small nodules <10 mm (Table 1).
Discussion
In the current study, we assessed the prevalence of adrenal nodular lesions and hyperplasia in patients with CD. We found that among 68 patients with CD who underwent adrenal imaging, 16 (23.5%) patients had an adrenal nodule ≥10 mm and 19 (27.9%) patients had adrenal hyperplasia or a nodule <10 mm. Additionally, we studied 10 patients with ACTH-dependent Cushing’s syndrome without tumor localization. Among these patients, 3 (30.0%) patients had an adrenal nodule ≥10 mm.
ACTH has trophic and mitogenic effects on the adrenal cortex [5], and chronic ACTH secretion may lead to adrenal hyperplasia. However, most of the literature on the co-existence of CD and adrenal nodules is based on case reports [16,17,18], with only a few studies focusing on the prevalence of adrenal nodules in cohorts of patients with CD [6, 7, 15].
Sohaib et al. reported that among 40 patients with CD, 25 (62.5%) had enlarged adrenal glands by CT, and seven patients (17.5% of the CD cohort) had an adrenal nodule ≥10 mm [7]. Imaki et al. found that among 24 patients with CD, 12 (50.0%) had adrenal hyperplasia, and only one (4.2%) had an adrenal nodule ≥10 mm [6]. Albiger et al. also studied the prevalence of adrenal nodules in CD, but defined a nodule as ≥5 mm. In their study, 15 out of 41 patients (36.6%) had adrenal nodules of this size [15] (Table 4). To the best of our knowledge, our study represents the largest investigation to date on the prevalence of adrenal abnormalities in CD patients (Table 4).
Table 4 Adrenal morphology in CD patients in the current study and three other main cohorts
In our study all patients underwent adrenal imaging during the active phase of their disease. Notably, 28 (41.2%) patients underwent adrenal imaging as part of their medical evaluation following the diagnosis of Cushing’s syndrome or due to persistent or recurrent disease. Shoaib et al. included CD patients who underwent CT imaging as part of their radiological assessment [7]. Albiger et al. included CD patients who had undergone abdominal CT scan as part of their initial evaluation or the assessment for persistent or recurrent disease [15].
With the increased use of abdominal imaging in recent decades, incidental findings of abnormal adrenal lesions have become more common [8]. The prevalence of AIs in the general poppulation has been reported to range from 1.2–5.0% in various studies [8, 10,11,12,13,14, 19], which is significantly lower than the prevalence observed in our CD patients.
A large retrospective cohort from the United Kingdom, including 479,975 outpatients that underwent in-hospital CT or MRI scans (excluding patients with known adrenal lesion), found that 1.2% of individuals had AI [19]. The prevalence of AI was higher in patients who underwent abdominal CT imaging, reaching to 3.0%. The authors found a correlation between age and the prevalence of AI, ranging from 0.2% in the youngest group (21–30 years) to 4.1% in the oldest group (age ≥91 years) [19]. Consistent with findings from previous studies [7, 15], our study also demonstrates that patients with adrenal nodules were significantly older compared to those with normal adrenal glands.
A recent study from China, which examined 25,356 healthy individuals (unselected population) who underwent abdominal CT imaging, found that 351 (1.4%) had an adrenal tumor [13]. Compared to the results of this study, we observed that in each age group, CD patients had a much higher prevalence of adrenal nodules: between 26–35 years 16.7% in our cohort vs 0.3% in the Chinese population, between 36–45 years 27.8% vs 0.7%, for subjects 46–55 years 30.8% vs 1.6%, and above the age of 55 years 26.3% vs 2.5% (Fig. 3).
We found that patients with adrenal nodules≥10 mm had a significantly lower ACTH level. Albiger et al. also found that ACTH levels were significantly lower in patients with adrenal nodules. Their hypothesis was that a gradual transition from pituitary to adrenal autonomy might suppress ACTH production [15]. Previous reports have suggested that, in a subgroup of patients with prolonged ACTH stimulation, there might be a transition from pituitary dependent to adrenal dependent Cushing’s syndrome [20, 21]. Tabarin et al., found that dexamethasone suppressibility and the stimulatory effect of metyrapone on ACTH secretion were less in CD patients with hyperplasia and adrenal nodules than in those with diffuse adrenal hyperplasia, suggesting a greater degree of adrenal autonomy in the former [22]. Dalmazi et al., found somatic mutations in the gene encoding the catalytic α (Cα) subunit of protein kinase A (PKA; PRKACA) in adrenal nodules of two patients with long-standing CD [23]. PRKACA somatic mutations are the most common genetic finding in adrenal adenomas associated with ACTH-independent Cushing syndrome [24], therefore these genetic alterations could represent a possible mechanism underlying adrenal nodule formation and autonomous cortisol hyperproduction in a subgroup of patients with long-standing CD.
In our study, 4 patients with adrenal nodules underwent unilateral adrenalectomy, two of them achieved full remission after the surgery. Interestingly, Dalmazi et al., described also that a patient who underwent unilateral adrenalectomy of a 35 mm adrenal nodule, achieved clinical and biochemical remission [23]. It may be worthwhile to consider unilateral adrenalectomy in selected CD patients with persistent hypercortisolemia after TSS, who have a large unilateral adrenal nodule.
There are several limitations to this study, primarily due to its retrospective design. The study cohort consists of patients who were referred for follow-up at endocrinology departments within tertiary hospitals. This setting likely leads to a higher frequency of imaging studies compared to the general population, potentially leading to a higher rate of incidental adrenal findings. Most of the patients did not have repeated adrenal imaging during their follow-up, so it is not possible to assess whether there was a change in the appearance of the adrenal glands following disease remission.
Moreover, there were instances where sufficient clinical data was unavailable to definitively confirm a diagnosis of CD in some patients. Thus, 10 patients were excluded from the main cohort analysis.
Another limitation is the relatively small sample size of the study population, which resulted in some findings not reaching statistical significance.
In conclusion, our study found that abnormal adrenal imaging was present in 51.5% of patients with CD. Notably, the prevalence of adrenal nodules in our cohort is 10 times higher than in the general population across all age groups, emphasizing a marked difference of adrenal morphology between CD patients and healthy individuals and suggesting that chronic ACTH stimulation leads to adrenal nodule development. The relative low levels of ACTH in patients with adrenal nodules may reflect partial autonomous cortisol secretion in some adrenal nodules. In light of this, adrenal nodules in patients with CD appear to be a relatively common finding, highlighting the importance of thorough laboratory and imaging diagnosis to identify the cause of hypercortisolism.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
M. Fleseriu, R. Auchus, I. Bancos et al. Consensus on diagnosis and management of Cushing’s disease: a guideline update. Lancet Diabetes Endocrinol. 9(12), 847–875 (2021). https://doi.org/10.1016/S2213-8587(21)00235-7
A.P. Coll, B.G. Challis, G.S.H. Yeo et al. The effects of proopiomelanocortin deficiency on murine adrenal development and responsiveness to adrenocorticotropin. Endocrinology 145(10), 4721–4727 (2004). https://doi.org/10.1210/en.2004-0491
D. Chida, S. Nakagawa, S. Nagai et al. Melanocortin 2 receptor is required for adrenal gland development, steroidogenesis, and neonatal gluconeogenesis. Proc. Natl Acad. Sci. USA 104(46), 18205–18210 (2007). https://doi.org/10.1073/pnas.0706953104
T. Imaki, M. Naruse, K. Takano, Adrenocortical hyperplasia associated with ACTH-dependent Cushing’s syndrome: comparison of the size of adrenal glands with clinical and endocrinological data. Endocr. J. 51(1), 89–95 (2004). https://doi.org/10.1507/endocrj.51.89
S.A. Sohaib, J.A. Hanson, J.D. Newell-Price et al. CT appearance of the adrenal glands in adrenocorticotrophic hormone-dependent Cushing’s syndrome. AJR Am. J. Roentgenol. 172(4), 997–1002 (1999). https://doi.org/10.2214/ajr.172.4.10587135
M. Fassnacht, S. Tsagarakis, M. Terzolo et al. European Society of Endocrinology clinical practice guidelines on the management of adrenal incidentalomas, in collaboration with the European Network for the Study of Adrenal Tumors. Eur. J. Endocrinol. 189(1), G1–G42 (2023). https://doi.org/10.1093/ejendo/lvad066
L. Hammarstedt, A. Muth, B. Wängberg et al. Adrenal lesion frequency: a prospective, cross-sectional CT study in a defined region, including systematic re-evaluation. Acta Radiol. 51(10), 1149–1156 (2010). https://doi.org/10.3109/02841851.2010.516016
L. Barzon, N. Sonino, F. Fallo, G. Palu, M. Boscaro, Prevalence and natural history of adrenal incidentalomas. Eur. J. Endocrinol. 149(4), 273–285 (2003). https://doi.org/10.1530/eje.0.1490273
S. Bovio, A. Cataldi, G. Reimondo et al. Prevalence of adrenal incidentaloma in a contemporary computerized tomography series. J. Endocrinol. Invest. 29(4), 298–302 (2006). https://doi.org/10.1007/BF03344099
Y. Jing, J. Hu, R. Luo et al. Prevalence and characteristics of adrenal tumors in an unselected screening population: a cross-sectional study. Ann. Intern. Med. 175(10), 1383–1391 (2022). https://doi.org/10.7326/M22-1619
J.H. Song, F.S. Chaudhry, W.W. Mayo-Smith, The incidental adrenal mass on CT: prevalence of adrenal disease in 1,049 consecutive adrenal masses in patients with no known malignancy. AJR Am. J. Roentgenol. 190(5), 1163–1168 (2008). https://doi.org/10.2214/AJR.07.2799
N.M. Albiger, G. Occhi, F. Sanguin et al. Adrenal nodules in patients with Cushing’s disease: prevalence, clinical significance and follow-up. J. Endocrinol. Invest. 34(8), e204–e209 (2011). https://doi.org/10.3275/7349
G. Borretta, M. Terzolo, F. Cesario, I. Meineri, A. Pia, A. Angeli, Coexistence of unilateral adrenal macronodule and Cushing’s disease. Report of two cases. J. Endocrinol. Invest. 19(2), 131–135 (1996). https://doi.org/10.1007/BF03349849
M.K.M. Shakir, I.C. Ebrahim, A. Spiro, V.Q. Mai, T.D. Hoang, Coexistence of Cushing disease with a solitary adrenocorticotrophic hormone-dependent adrenal adenoma. AACE Clin. Case Rep. 8(1), 41–44 (2022). https://doi.org/10.1016/j.aace.2020.11.027
F.W.F. Hanna, S. Hancock, C. George et al. Adrenal incidentaloma: prevalence and referral patterns from routine practice in a large UK university teaching hospital. J. Endocr. Soc. 6(1), bvab180 (2022). https://doi.org/10.1210/jendso/bvab180
H.J.L.M. Timmers, E.M. van Ginneken, P. Wesseling, C.G.J. Sweep, A.R.M.M. Hermus, A patient with recurrent hypercortisolism after removal of an ACTH-secreting pituitary adenoma due to an adrenal macronodule. J. Endocrinol. Invest. 29(10), 934–939 (2006). https://doi.org/10.1007/BF03349200
J. Santos, I. Paiva, L. Gomes et al. [Recurrent hypercortisolism after removal of an ACTH secretor pituitary adenoma associated with an adrenal macronodule]. Acta Med. Port. 23(1), 107–112 (2010)
A. Tabarin, S. Magimel, F. Laurent, A. Navarranne, J. Guérin, P. Roger, [Biological and developmental aspects of macronodular adrenal hyperplasia in Cushing’s disease]. Ann. Endocrinol. 53(2), 59–66 (1992)
G. Di Dalmazi, H.J.L.M. Timmers, G. Arnaldi et al. Somatic PRKACA mutations: association with transition from pituitary-dependent to adrenal-dependent cushing syndrome. J. Clin. Endocrinol. Metab. 104(11), 5651–5657 (2019). https://doi.org/10.1210/jc.2018-02209
K. Bathon, I. Weigand, J.T. Vanselow et al. Alterations in protein kinase A substrate specificity as a potential cause of cushing syndrome. Endocrinology 160(2), 447–459 (2019). https://doi.org/10.1210/en.2018-00775
Open access funding provided by Tel Aviv University.
Author information
Authors and Affiliations
Endocrine and Diabetes Institute, Shamir Medical Center, Zerifin, Be’er Ya’akov, Israel
Efrat Markus & Shlomit Koren
Gray Faculty of Medical & Health Sciences, Tel Aviv University, Tel Aviv, Israel
Efrat Markus, Yaron Rudman, Shlomit Koren & Ilan Shimon
Institute of Endocrinology, Beilinson Hospital, Rabin Medical Center, Petah Tikva, Israel
Yaron Rudman & Ilan Shimon
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Efrat Markus, Yaron Rudman, and Ilan Shimon. The first draft of the manuscript was written by Efrat Markus and Ilan Shimon and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.






 Accepted manuscripts
Accepted manuscripts