Medical therapy for Cushing’s syndrome (CS) is increasingly used. A potent adrenal steroidogenesis inhibitor, osilodrostat, has been rarely linked to prolonged adrenal insufficiency (AI).
Objective
We hypothesized that osilodrostat-induced adrenal insufficiency could be associated with adrenal gland shrinkage.
Design
Non-interventional, retrospective, longitudinal, IRB-approved study of patients with CS treated at Oregon Health and Science University between January 1, 2000 and July 1, 2025.
Setting
Ambulatory and inpatient, academic, quaternary medical center.
Patients or Other Participants
Patients with ACTH-dependent CS, treated with osilodrostat for >3 months, and CT imaging before and after osilodrostat available for adrenal volume (AV) measurement.
Intervention(s)
Age, sex, osilodrostat doses and duration, laboratory data and AI were recorded. AV was calculated using manual segmentation on CT images by a board-certified radiologist.
Main Outcome Measure(s)
AV before and after initiation of osilodrostat was expressed as percent reduction.
Results
10 patients (5 ectopic CS, 4 unknown ACTH source, 1 Cushing’s disease) were included. Osilodrostat mean starting, maximum and final doses: 7.7, 13.8 and 5.9 mg/day, respectively, over 23 months. Four patients received block-and-replace regimen, AI developed in 5. Adrenal gland volume decreased by 46.7±22.2% from 25.5±9.9 ml to 12.7±6.4 ml, p<0.001 over a median of 19 months. AV reduction positively correlated with maximum osilodrostat dose, r=0.626, p=0.027.
Conclusions
We found that in selected patients with ACTH-dependent CS, osilodrostat can induce significant adrenal shrinkage, with or without AI. Further confirmation by larger studies of different CS types and monitoring for AI is required for all patients.
Up to 50% of patients with adrenal incidentalomas have mild autonomous cortisol secretion, which may increase their cardiometabolic morbidity, compared with patients with nonfunctional adrenal tumors. Studies evaluating cardiometabolic outcomes of patients with mild autonomous cortisol secretion defined by 1-mg dexamethasone suppression testing (cortisol 1.8–5 μg/dL) have demonstrated mixed results. The aim of this study was to assess the metabolic outcomes of patients with mild autonomous cortisol secretion, defined by the 1-mg dexamethasone suppression testing criterion, compared with patients with nonfunctional adrenal tumors who underwent adrenalectomy.
Methods
We conducted a single-institution retrospective cohort study comparing adult patients who underwent unilateral adrenalectomy from November 30, 2011, to August 19, 2023, for mild autonomous cortisol secretion (1-mg dexamethasone suppression testing cortisol 1.8–5 μg/dL) or nonfunctional adrenal tumors (1-mg dexamethasone suppression testing cortisol <1.8 μg/dL). Preoperative prevalences and postoperative changes in diabetes mellitus, hypertension, dyslipidemia, and elevated body mass index (≥25) were assessed. Patients were followed from the time of surgery until their last outpatient visit. Multivariable logistic regression was pursued for outcomes that varied between cohorts.
Results
A total of 65 patients (53 mild autonomous cortisol secretion and 12 nonfunctional adrenal tumors) were analyzed. Patients with mild autonomous cortisol secretion were older and more likely to have diabetes mellitus than patients with nonfunctional adrenal tumors (odds ratio: 7.81, 95% confidence interval [0.94, 64.96], P = .04). Patients were followed for a median of 28.1 months [11.1, 55.3 months]. Patients with mild autonomous cortisol secretion were more likely to have postoperative weight improvement (odds ratio: 8.31, [0.97, 71.14], P = .03). After adjusting for clinically relevant variables, the 1-mg dexamethasone suppression testing cortisol was predictive of postoperative weight improvement (odds ratio: 1.88, [1.1, 3.65], P = .04).
Conclusion
Weight loss should be considered as a potential benefit of adrenalectomy in patients with mild autonomous cortisol secretion.
Mild autonomous cortisol secretion (MACS) is the most common hormonal abnormality diagnosed in patients with adrenal incidentalomas, impacting 20%–50% of patients.1 Patients with MACS have biochemical evidence of adrenocorticotropic hormone (ACTH)-independent hypercortisolism but lack clinical stigmata commonly associated with overt hypercortisolism, such as facial plethora, abdominal adiposity, extremity weakness and wasting, and/or violaceous striae.2 Overt hypercortisolism is well recognized to cause cardiovascular, musculoskeletal, and metabolic disorders, which have variable resolution even after diagnosis and treatment.3 There is a growing body of evidence that patients with MACS also have increased cardiometabolic morbidity and mortality compared with patients with nonfunctional adrenal tumors,4 but this evidence is challenging to interpret given wide variability in diagnostic criteria that have historically been used.5, 6, 7
Recent guidelines have suggested that a diagnosis of MACS be applied to all patients with a morning (AM) serum cortisol of >1.8 μg/dL after low-dose (1-mg) dexamethasone suppression testing (DST) who lack overt features of hypercortisolism.8,9 However, prior studies comparing cardiometabolic outcomes between patients with MACS and nonfunctional adrenal tumors as well as between patients who underwent operative and nonoperative management have used a 1-mg DST AM serum cortisol of 1.8–5.0 μg/dL as a definition of mild (“subclinical”) hypercortisolism.10, 11, 12, 13, 14, 15, 16 Given that these studies have demonstrated mixed results,4 the primary aim of this study was to assess the metabolic outcomes of patients with MACS, as defined by a 1-mg DST AM cortisol of 1.8–5.0 μg/dL, compared with patients with nonfunctional adrenal tumors who underwent adrenalectomy.
Section snippets
Methods
This was a single-institution retrospective cohort study of patients aged ≥18 years who underwent initial unilateral adrenalectomy from November 30, 2011, to August 19, 2023. Patients were identified through a prospectively maintained database of all patients who underwent adrenalectomy at the study institution. Patients were excluded if they had a 1-mg DST AM serum cortisol of >5 μg/dL, ACTH-dependent hypercortisolism, primary aldosteronism, pheochromocytoma, primary bilateral macronodular
Results
Of the 460 patients who underwent adrenalectomy during the study period, 53 patients met criteria for MACS and 12 patients for nonfunctional adrenal tumors, yielding a cohort of 65 patients. Patients with MACS were older than those with nonfunctional adrenal tumors (MACS, median 60 years [IQR: 54, 68 years] vs nonfunctional adrenal tumors, 49 years [37, 57 years], P = .02) but were similar by sex, race, ethnicity, BMI, nodule size, laterality, and surgical approach (Table II). Among patients
Discussion
MACS is the most common hormonal abnormality diagnosed in patients with adrenal incidentalomas. Despite lacking clinical stigmata of overt hypercortisolism, patients with MACS appear to have increased cardiometabolic morbidity and mortality similar to patients with overt hypercortisolism. The optimal management of MACS is debated, and prior studies using a 1-mg DST AM serum cortisol of 1.8–5.0 μg/dL as a definition of mild hypercortisolism have demonstrated mixed results. Hence, this study
Funding/Support
This project is funded in part by the Advancing a Healthier Wisconsin Endowment at the Medical College of Wisconsin. This publication was supported by the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health (NIH), through grant number UL1TR001436. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. The grant supports the creation and maintenance of
CRediT authorship contribution statement
Alexa Lisevick Kumar: Writing – original draft, Visualization, Methodology, Formal analysis, Data curation, Conceptualization. Sophie Dream: Writing – review & editing, Validation, Supervision, Methodology, Investigation. Tahseen Shaik: Resources, Project administration, Investigation, Data curation. Kara Doffek: Resources, Project administration, Investigation, Data curation. Ryan Conrardy: Writing – review & editing, Methodology, Formal analysis. James W. Findling: Writing – review & editing,
Conflict of Interest/Disclosure
Dr Findling reports consulting for Corcept, Diurnal, Crinetics and serving as an investigator for Recordati. The rest of the authors reported no biomedical financial interests or potential conflicts of interest.
Mild autonomous cortisol secretion: pathophysiology, comorbidities and management approaches
Nat Rev Endocrinol
(2024)
L.K. Nieman
Cushing’s syndrome: update on signs, symptoms and biochemical screening
Eur J Endocrinol
(2015)
S. Puglisi et al.
Long-term consequences of cushing syndrome: a systematic literature review
J Clin Endocrinol Metab
(2024)
I.C.M. Pelsma et al.
Comorbidities in mild autonomous cortisol secretion and the effect of treatment: systematic review and meta-analysis
Eur J Endocrinol
(2023)
X. Ren et al.
Evaluating the efficacy of surgical and conservative approaches in mild autonomous cortisol secretion: a meta-analysis
Front Endocrinol (Lausanne)
(2024)
M.M. Khadembashiri et al.
Comparison of adrenalectomy with conservative treatment on mild autonomous cortisol secretion: a systematic review and meta-analysis
Front Endocrinol
(2024)
Y.S. Elhassan et al.
Natural history of adrenal incidentalomas with and without mild autonomous cortisol excess
Ann Intern Med
(2019)
M. Fassnacht et al.
European society of endocrinology clinical practice guidelines on the management of adrenal incidentalomas, in collaboration with the European network for the study of adrenal tumors
Eur J Endocrinol
(2023)
L. Yip et al.
American association of endocrine surgeons guidelines for adrenalectomy: executive summary
New data on relacorilant (Corcept Therapeutics), a selective glucocorticoid receptor modulator, revealed several cardiometabolic benefits for patients with hypercortisolism.
Researchers presented results from the GRACE and GRADIENT trials, which assessed relacorilant in adults with hypercortisolism. GRACE was an open-label trial that enrolled adults with endogenous hypercortisolism, whereas GRADIENT included those with adrenal hypercortisolism and randomly assigned participants to relacorilant or placebo.
Both trials demonstrated similar reductions in body weight. The relacorilant group in GRADIENT had a 3.6 kg reduction in body weight, and adults in GRACE reduced their body weight by 3.3 kg at 22 weeks.
“Relacorilant may improve many of the common features of hypercortisolism, which may provide a holistic benefit to our patients,” Oksana Hamidi, DO, MSCS, study investigator and associate professor in the division of endocrinology at UT Southwestern Medical Center, told Healio | Endocrine Today. “An interesting observation was that relacorilant can lead to weight loss, and that weight loss is mostly fat mass, with lean mass being preserved or even increasing. The ability to maintain muscle is particularly important for our patients.”
In a cardiometabolic analysis, adults with hypertension receiving relacorilant had greater reductions in both systolic and diastolic blood pressure compared with placebo. For adults with hyperglycemia at baseline, the relacorilant group had greater declines in fasting glucose and glucose area under the curve.
Corin Badiu, MD, study investigator, professor of endocrinology and head of the department of endocrinology IV in the National Institute of Endocrinology and “C.Davila” University of Medicine and Pharmacy in Bucharest, Romania, and fellow of the Romanian Academy of Medical Sciences, said the benefits of relacorilant may extend into additional areas that could be studied in the future.
“Apart from metabolic and cardiovascular improvements, we expect long-term improvements in bone mass, liver steatosis, mood, sleep and other behavioral aspects [that] are disturbed in hypercortisolism,” Badiu told Healio | Endocrine Today.
Irina Bancos, MD, MSc, professor of medicine in the division of endocrinology, metabolism and nutrition at Mayo Clinic, said relacorilant could provide benefits similar to mifepristone (Korlym, Corcept Therapeutics) for patients with hypercortisolism, but with fewer adverse events related to progesterone health. Bancos was not involved with the trial.
“Why is there a need for another medication in the same class by the same company? The major reason is to achieve the same metabolic impact as far as weight loss and improvement of hyperglycemia … but also to decrease the side effects,” Bancos told Healio | Endocrine Today.
IntroductionCushing’s disease (CD) is the most common cause of endogenous Cushing’s syndrome. Adrenocorticotropic hormone (ACTH) has trophic and mitogenic effects on the adrenal cortex that may cause diffuse adrenal enlargement and nodular lesions.
AimTo evaluate the prevalence of adrenal structural abnormalities in patients with CD.
MethodsRetrospective cohort study. We conducted a computerized search in our medical centers databases for the diagnosis of CD recorded between the years 1995–2024. Out of 124 patients with ACTH dependent Cushing’s syndrome, we identified 68 patients with CD who underwent adrenal imaging. We analyzed the clinical, biochemical, and imaging data.
ResultsOur cohort included 68 patients (51 females, 75.0%; mean age at the time of adrenal imaging, 44.6 ± 14.9 years). Sixteen (23.5%) patients had an adrenal nodule ≥10 mm (median size, 27.5 mm, IQR 14.3–38.3), and 19 others (27.9%) had adrenal hyperplasia or nodules <10 mm. The prevalence of adrenal nodules increased with age from 16.7% in patients aged 26–35 years to 26.3% in those aged above 55. Patients with adrenal nodules were older compared to those with normal adrenal glands (mean age, 49.0 ± 12.4 vs 39.1 ± 14.9 years; p = 0.03), and had lower ACTH level (0.7 x ULN, IQR 0.5–1.2, vs 1.2 x ULN, IQR 0.9–1.8, p = 0.02).
ConclusionsWe identified abnormal adrenal imaging in 51.5% of patients with CD. The prevalence of adrenal nodules in our study was 10-fold higher than in the normal population, for all age groups. This suggests that chronic ACTH secretion in CD is associated with adrenal nodules appearance.
Explore related subjects
Discover the latest articles, books and news in related subjects, suggested using machine learning.
Cushing’s disease (CD) is the most common etiology (70%) of endogenous Cushing’s syndrome [1]. CD is caused by a pituitary adenoma that autonomously secretes adrenocorticotropic hormone (ACTH), leading to cortisol overproduction and secretion from the adrenal cortex [2].
ACTH is a predominant trophic factor of the adrenal cortex. Several animal models, in which ACTH or its receptor (melanocortin 2 receptor, MC2R) were eliminated, have confirmed the central role of ACTH in maintaining normal growth of the adrenal cortex [3, 4]. While ACTH also exerts a mitogenic effect, the precise mechanism by which it promotes adrenocortical growth and proliferation is complex and only partially understood [5]. Due to the effects of ACTH on the adrenal cortex, patients with ACTH-dependent Cushing’s syndrome have high prevalence of adrenal hyperplasia, reaching up to 60% [6, 7].
Adrenal incidentaloma (AI) is an adrenal mass (≥1 cm) detected on imaging not performed for a suspected adrenal disease [8]. Autopsy studies suggest that the overall prevalence of adrenal masses ranges from 1.1–8.7%, which increases with age [9]. In recent decades, with advancement in imaging technologies, radiological studies have become more common and accurate, with the prevalence of AI’s in imaging studies approaching values similar to those found in autopsy studies [8, 10,11,12,13,14].
In contrast to adrenal hyperplasia, there are limited studies examining the prevalence of adrenal nodules in CD. These studies, which included small cohorts of up to 40 patients with CD, found a significantly higher prevalence of adrenal nodules, with rates ranging from 4% to nearly 40% among studied individuals [6, 7, 15].
Our aim was to study the morphology of the adrenal glands and assess the prevalence of abnormal adrenal findings, including hyperplasia and adrenal nodules, in a large cohort of patients with CD.
Materials and methods
We conducted a computerized search in Rabin and Shamir Medical Centers databases for the diagnosis of Cushing’s syndrome and screened for patients with ACTH-dependent Cushing’s syndrome. Cushing’s syndrome was diagnosed in patients with characteristic symptoms and signs and hypercortisolemia. Hypercortisolemia was confirmed according to laboratory findings including high 24 h urinary free cortisol (UFC), elevated midnight salivary cortisol, and abnormal 1 mg dexamethasone suppression test. We further classified these patients to ACTH-dependent Cushing’s syndrome, based on ACTH level in the normal or above the normal range.
A diagnosis of CD was confirmed by a pituitary adenoma of ≥6 mm depicted by seller MRI, an inferior petrosal sinus sampling (IPSS) supporting a pituitary source of ACTH secretion, immunostaining for ACTH and/or T-PIT in resected tumor specimens, and/or hormonal remission following successful trans-sphenoidal adenoma resection.
After identifying all patients with CD, our cohort included only patients with CD who have undergone abdominal computed tomography (CT) or abdominal magnetic resonance imaging (MRI), during the active phase of their disease.
In addition, we assembled a cohort of patients with ACTH-dependent Cushing’s syndrome in whom the source of ACTH secretion was not identified, that is, patients with ACTH-dependent Cushing’s syndrome without tumor localization.
Patients with malignant pituitary tumor and those with confirmed ectopic ACTH secretion were excluded. Patients that were treated with glucocorticoids were also excluded.
Based on an imaging report from an expert radiologist, all adrenal images were classified into three categories: normal, hyperplastic, or nodular adrenal glands. All adrenal nodules were classified according to nodule size: maximal nodule diameter below 10 mm, or ≥10 mm.
The study was approved by the Rabin Medical Center and Shamir Medical Center institutional review boards with waiver of patient consent, as complied with the Helsinki Declaration.
The authors received no funding for performing this study.
Statistical analysis and plan
Statistical analysis was performed using IBM SPSS version 29.0 (IBM Corp., Armonk, NY).
Continuous variables were presented by Mean (SD) or Median (IQR) as appropriate. Dichotomous variables were presented by N (%).
T-test and Mann–Whitney tests were used to compare values of normally and non-normally distributed continuous variables, with Chi-Square test used for comparison of categorical variables.
Two-sided P-values less than 0.05 were considered statistically significant.
Results
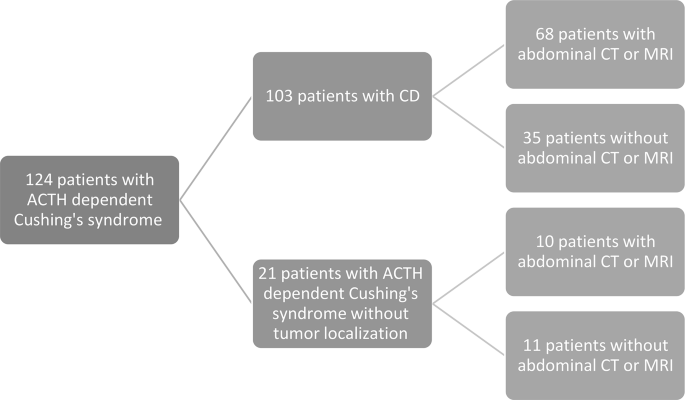
From March 1995 to December 2024, a total of 124 patients with ACTH-dependent Cushing’s syndrome were identified. There were 103 patients with the diagnosis of CD, and 21 patients with ACTH-dependent Cushing’s syndrome without tumor localization. After carefully reviewing each case, we excluded 35 patients without reported adrenal imaging (Fig. 1).
Fig. 1
Patient selection flowchart. ACTH adrenocorticotropic hormone, CD Cushing’s disease, CT computed tomography, MRI magnetic resonance imaging
The main cohort included 68 patients with CD and available adrenal imaging (51 females, 75.0%; mean age at the time of adrenal imaging, 44.6 ± 14.9 years). The median pituitary adenoma size (i.e. largest adenoma diameter) was 6 mm (IQR 4.75–10.25). The median ACTH level was 1.2 x ULN (IQR 0.8–1.9), and the median UFC level was 3.5 x ULN (IQR 2.0–6.0) (Table 1).
Table 1 Baseline characteristics of 68 patients with Cushing’s disease and 10 subjects with ACTH dependent Cushing’s syndrome without tumor localization
Sixty-five (95.6%) patients underwent trans-sphenoidal surgery (TSS), 44 of them (67.7%) had a resected adenoma expressing ACTH and/or T-PIT, 7 (10.8%) had no pituitary adenoma in pathology, and for 14 (21.5%) patients we did not have available pathology reports. Forty-six of the 65 (70.8%) patients experienced hormonal remission following surgery (Table 1).
During follow-up, 17 (26.2%) patients underwent repeated pituitary surgery, 8 of them due to persistent disease, and 9 due to recurrent elevated cortisol. Fifteen (22.1%) patients underwent radiation therapy. Thirty-nine (57.4%) patients received medical therapy for CD with adrenal steroidogenesis inhibitors and medications targeting pituitary somatostatin or dopamine receptors. Of these, seven (17.9%) patients were treated medically before TSS, 31 (79.5%) after TSS, and one (2.6%) patient received medical therapy both before and after TSS (Table 1).
Abdominal imaging findings in patients with CD
Sixty-three patients had abdominal CT, including five who had Ga68 positron emission tomography (PET) CT without pathologic adrenal Ga68 uptake on functional imaging. Five additional patients had abdominal MRI.
Twenty-three (33.8%) patients underwent adrenal imaging as part of their medical evaluation following the diagnosis of Cushing’s syndrome. Five (7.4%) patients underwent adrenal imaging due to persistent or recurrent disease following surgery. Eleven (16.2%) patients had adrenal imaging because of abdominal pain, thirteen (19.1%) patients for other reasons not related to Cushing’s syndrome (e.g. following abnormal finding in ultrasonography (US), or before abdominal/gynecological surgery) and sixteen patients (23.5%) for unknown reasons.
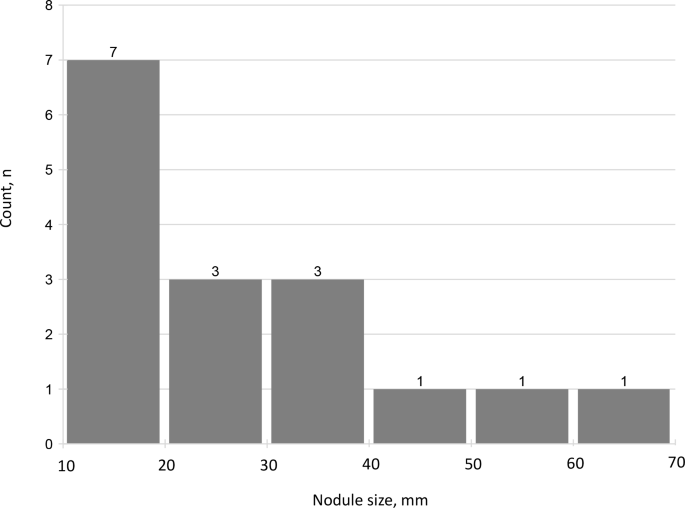
A total of 16 (23.5%) patients had an adrenal nodule ≥10 mm (median size, 27.5 mm, IQR 14.3–38.3) (Table 1). All of the nodules were radiologically defined as compatible with adenomas, based on low Hounsfield Units (HU) on non-contrast CT or signal drop on out-of-phase MRI sequences. Five patients had nodules in the right adrenal gland, six others had nodules in the left adrenal gland, and five patients had adrenal nodules ≥10 mm in both adrenal glands – one nodule in each side. There was only one patient with adrenal imaging consistent with bilateral multinodular adrenal hyperplasia, that was classified into the group of patients with adrenal nodule ≥10 mm. Nine (13.2%) patients had an adrenal nodule ≥20 mm (Fig. 2).
Nineteen (27.9%) patients had adrenal hyperplasia and/or nodules smaller than 10 mm (Table 1).
Patients with adrenal nodules ≥10 mm, as well as those with adrenal hyperplasia and/or nodules <10 mm, were significantly older compared to individuals with normal adrenal glands (mean age at imaging: 49.0 ± 12.4 and 50.4 ± 13.7 vs 39.1 ± 14.9 years, respectively; p = 0.03 and p = 0.01). Only patients with adrenal nodules ≥10 mm had significantly lower ACTH levels compared to patients with normal adrenal glands (0.7 x ULN, IQR 0.5–1.2, vs 1.2 x ULN, IQR 0.9–1.8; p = 0.02) (Table 2).
Table 2 Clinical characteristics of CD patients with adrenal nodules ≥10 mm, adrenal hyperplasia and/or nodule <10mm and with normal adrenal glands
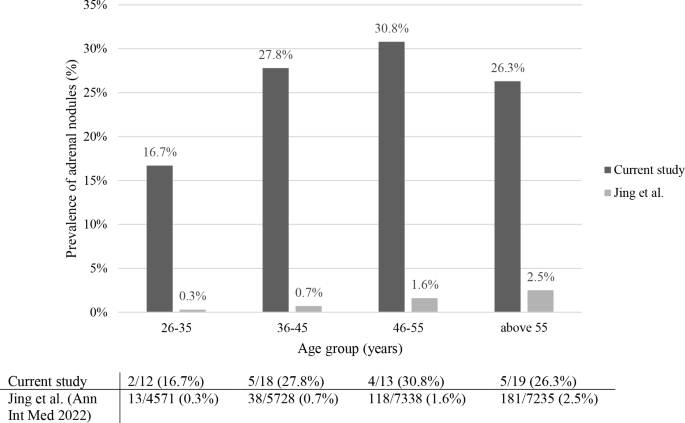
The prevalence of adrenal nodules increased with age from 16.7% in patients aged 26–35 years to 27.8, 30.8 and 26.3% in those aged 36–45, 46–55, and above 55 years, respectively (Fig. 3).
Fig. 3
Prevalence of adrenal nodules ≥10 mm in CD patients (current study) and in the general population (Jing et al.), stratified by age group
Urinary free cortisol (UFC) levels did not differ significantly among the three groups. Patients with adrenal nodules ≥10 mm had a median UFC level of 4.0 x ULN (IQR 1.7–6.3), those with adrenal hyperplasia and/or nodules <10 mm had a median of 3.8 x ULN (IQR 3.0–6.0), and patients with normal adrenal glands had a median of 3.5 x ULN (IQR 2.0–6.0) (p = 0.77 and p = 0.63, respectively vs. normal adrenal glands) (Table 2).
A pituitary adenoma was depicted by sellar MRI in 54 (79.4%) patients. Patients with adrenal nodules ≥10 mm tended to harbor a larger pituitary adenoma compared to patients with normal adrenals (median adenoma size, 7.0 mm (IQR 6.0–11.5) vs 6.0 mm (IQR 4.0–8.0) respectively; p = 0.11). Patients with adrenal hyperplasia and/or nodules <10 mm had a median adenoma size of 5.5 mm (IQR 3.0–13.0), which was not significantly different compared to patients with normal adrenal glands (p = 0.63) (Table 2).
Remission rates after TSS were not significantly different among the groups. Among patients with adrenal nodules ≥10 mm, 12 (80%) patients achieved remission, compared to 23 (71.9%) patients with normal adrenal glands (p = 0.73). Similarly, 11 patients (61.1%) with adrenal hyperplasia and/or nodules <10 mm achieved remission, with no significant difference compared to patients with normal adrenal glands (p = 0.53) (Table 2).
Clinical characteristics of four patients with CD who underwent unilateral adrenalectomy
Four patients (all females; mean age at the time of adrenal imaging, 46.3 ± 6.8 years) underwent unilateral adrenalectomy for a benign adrenocortical adenoma (adrenal adenoma size between 2.8–6.4 cm). One patient underwent adrenalectomy prior to TSS, with persistent hypercortisolism following adrenal surgery but achieved remission after TSS (Table 3).
Table 3 Clinical characteristics of four patients with CD who underwent unilateral adrenalectomy
Three patients underwent adrenalectomy after TSS. All three patients exhibited persistent hypercortisolism prior to adrenalectomy, with a median UFC level of 3.0 x ULN (IQR 1.9–6.1). The median ACTH level was 0.6 x ULN (IQR 0.3–1.1). One of them experienced transient postoperative cortisol normalization, while the other two achieved remission following adrenalectomy (Table 3).
Abdominal imaging findings in patients with ACTH-dependent Cushing’s syndrome without tumor localization
Ten patients (including 7 females) with ACTH-dependent Cushing’s syndrome without tumor localization had adrenal imaging. Compared to patients with CD, these patients were older (mean age at the time of adrenal imaging, 58.4 ± 15.1 vs 44.7 ± 14.9 years, p < 0.01). Three of these patients (30.0%) had adrenal nodule ≥10 mm (median size, 14 mm, IQR 11.7–18), and 6 (60.0%) had adrenal hyperplasia or small nodules <10 mm (Table 1).
Discussion
In the current study, we assessed the prevalence of adrenal nodular lesions and hyperplasia in patients with CD. We found that among 68 patients with CD who underwent adrenal imaging, 16 (23.5%) patients had an adrenal nodule ≥10 mm and 19 (27.9%) patients had adrenal hyperplasia or a nodule <10 mm. Additionally, we studied 10 patients with ACTH-dependent Cushing’s syndrome without tumor localization. Among these patients, 3 (30.0%) patients had an adrenal nodule ≥10 mm.
ACTH has trophic and mitogenic effects on the adrenal cortex [5], and chronic ACTH secretion may lead to adrenal hyperplasia. However, most of the literature on the co-existence of CD and adrenal nodules is based on case reports [16,17,18], with only a few studies focusing on the prevalence of adrenal nodules in cohorts of patients with CD [6, 7, 15].
Sohaib et al. reported that among 40 patients with CD, 25 (62.5%) had enlarged adrenal glands by CT, and seven patients (17.5% of the CD cohort) had an adrenal nodule ≥10 mm [7]. Imaki et al. found that among 24 patients with CD, 12 (50.0%) had adrenal hyperplasia, and only one (4.2%) had an adrenal nodule ≥10 mm [6]. Albiger et al. also studied the prevalence of adrenal nodules in CD, but defined a nodule as ≥5 mm. In their study, 15 out of 41 patients (36.6%) had adrenal nodules of this size [15] (Table 4). To the best of our knowledge, our study represents the largest investigation to date on the prevalence of adrenal abnormalities in CD patients (Table 4).
Table 4 Adrenal morphology in CD patients in the current study and three other main cohorts
In our study all patients underwent adrenal imaging during the active phase of their disease. Notably, 28 (41.2%) patients underwent adrenal imaging as part of their medical evaluation following the diagnosis of Cushing’s syndrome or due to persistent or recurrent disease. Shoaib et al. included CD patients who underwent CT imaging as part of their radiological assessment [7]. Albiger et al. included CD patients who had undergone abdominal CT scan as part of their initial evaluation or the assessment for persistent or recurrent disease [15].
With the increased use of abdominal imaging in recent decades, incidental findings of abnormal adrenal lesions have become more common [8]. The prevalence of AIs in the general poppulation has been reported to range from 1.2–5.0% in various studies [8, 10,11,12,13,14, 19], which is significantly lower than the prevalence observed in our CD patients.
A large retrospective cohort from the United Kingdom, including 479,975 outpatients that underwent in-hospital CT or MRI scans (excluding patients with known adrenal lesion), found that 1.2% of individuals had AI [19]. The prevalence of AI was higher in patients who underwent abdominal CT imaging, reaching to 3.0%. The authors found a correlation between age and the prevalence of AI, ranging from 0.2% in the youngest group (21–30 years) to 4.1% in the oldest group (age ≥91 years) [19]. Consistent with findings from previous studies [7, 15], our study also demonstrates that patients with adrenal nodules were significantly older compared to those with normal adrenal glands.
A recent study from China, which examined 25,356 healthy individuals (unselected population) who underwent abdominal CT imaging, found that 351 (1.4%) had an adrenal tumor [13]. Compared to the results of this study, we observed that in each age group, CD patients had a much higher prevalence of adrenal nodules: between 26–35 years 16.7% in our cohort vs 0.3% in the Chinese population, between 36–45 years 27.8% vs 0.7%, for subjects 46–55 years 30.8% vs 1.6%, and above the age of 55 years 26.3% vs 2.5% (Fig. 3).
We found that patients with adrenal nodules≥10 mm had a significantly lower ACTH level. Albiger et al. also found that ACTH levels were significantly lower in patients with adrenal nodules. Their hypothesis was that a gradual transition from pituitary to adrenal autonomy might suppress ACTH production [15]. Previous reports have suggested that, in a subgroup of patients with prolonged ACTH stimulation, there might be a transition from pituitary dependent to adrenal dependent Cushing’s syndrome [20, 21]. Tabarin et al., found that dexamethasone suppressibility and the stimulatory effect of metyrapone on ACTH secretion were less in CD patients with hyperplasia and adrenal nodules than in those with diffuse adrenal hyperplasia, suggesting a greater degree of adrenal autonomy in the former [22]. Dalmazi et al., found somatic mutations in the gene encoding the catalytic α (Cα) subunit of protein kinase A (PKA; PRKACA) in adrenal nodules of two patients with long-standing CD [23]. PRKACA somatic mutations are the most common genetic finding in adrenal adenomas associated with ACTH-independent Cushing syndrome [24], therefore these genetic alterations could represent a possible mechanism underlying adrenal nodule formation and autonomous cortisol hyperproduction in a subgroup of patients with long-standing CD.
In our study, 4 patients with adrenal nodules underwent unilateral adrenalectomy, two of them achieved full remission after the surgery. Interestingly, Dalmazi et al., described also that a patient who underwent unilateral adrenalectomy of a 35 mm adrenal nodule, achieved clinical and biochemical remission [23]. It may be worthwhile to consider unilateral adrenalectomy in selected CD patients with persistent hypercortisolemia after TSS, who have a large unilateral adrenal nodule.
There are several limitations to this study, primarily due to its retrospective design. The study cohort consists of patients who were referred for follow-up at endocrinology departments within tertiary hospitals. This setting likely leads to a higher frequency of imaging studies compared to the general population, potentially leading to a higher rate of incidental adrenal findings. Most of the patients did not have repeated adrenal imaging during their follow-up, so it is not possible to assess whether there was a change in the appearance of the adrenal glands following disease remission.
Moreover, there were instances where sufficient clinical data was unavailable to definitively confirm a diagnosis of CD in some patients. Thus, 10 patients were excluded from the main cohort analysis.
Another limitation is the relatively small sample size of the study population, which resulted in some findings not reaching statistical significance.
In conclusion, our study found that abnormal adrenal imaging was present in 51.5% of patients with CD. Notably, the prevalence of adrenal nodules in our cohort is 10 times higher than in the general population across all age groups, emphasizing a marked difference of adrenal morphology between CD patients and healthy individuals and suggesting that chronic ACTH stimulation leads to adrenal nodule development. The relative low levels of ACTH in patients with adrenal nodules may reflect partial autonomous cortisol secretion in some adrenal nodules. In light of this, adrenal nodules in patients with CD appear to be a relatively common finding, highlighting the importance of thorough laboratory and imaging diagnosis to identify the cause of hypercortisolism.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
M. Fleseriu, R. Auchus, I. Bancos et al. Consensus on diagnosis and management of Cushing’s disease: a guideline update. Lancet Diabetes Endocrinol. 9(12), 847–875 (2021). https://doi.org/10.1016/S2213-8587(21)00235-7
A.P. Coll, B.G. Challis, G.S.H. Yeo et al. The effects of proopiomelanocortin deficiency on murine adrenal development and responsiveness to adrenocorticotropin. Endocrinology 145(10), 4721–4727 (2004). https://doi.org/10.1210/en.2004-0491
D. Chida, S. Nakagawa, S. Nagai et al. Melanocortin 2 receptor is required for adrenal gland development, steroidogenesis, and neonatal gluconeogenesis. Proc. Natl Acad. Sci. USA 104(46), 18205–18210 (2007). https://doi.org/10.1073/pnas.0706953104
T. Imaki, M. Naruse, K. Takano, Adrenocortical hyperplasia associated with ACTH-dependent Cushing’s syndrome: comparison of the size of adrenal glands with clinical and endocrinological data. Endocr. J. 51(1), 89–95 (2004). https://doi.org/10.1507/endocrj.51.89
S.A. Sohaib, J.A. Hanson, J.D. Newell-Price et al. CT appearance of the adrenal glands in adrenocorticotrophic hormone-dependent Cushing’s syndrome. AJR Am. J. Roentgenol. 172(4), 997–1002 (1999). https://doi.org/10.2214/ajr.172.4.10587135
M. Fassnacht, S. Tsagarakis, M. Terzolo et al. European Society of Endocrinology clinical practice guidelines on the management of adrenal incidentalomas, in collaboration with the European Network for the Study of Adrenal Tumors. Eur. J. Endocrinol. 189(1), G1–G42 (2023). https://doi.org/10.1093/ejendo/lvad066
L. Hammarstedt, A. Muth, B. Wängberg et al. Adrenal lesion frequency: a prospective, cross-sectional CT study in a defined region, including systematic re-evaluation. Acta Radiol. 51(10), 1149–1156 (2010). https://doi.org/10.3109/02841851.2010.516016
L. Barzon, N. Sonino, F. Fallo, G. Palu, M. Boscaro, Prevalence and natural history of adrenal incidentalomas. Eur. J. Endocrinol. 149(4), 273–285 (2003). https://doi.org/10.1530/eje.0.1490273
S. Bovio, A. Cataldi, G. Reimondo et al. Prevalence of adrenal incidentaloma in a contemporary computerized tomography series. J. Endocrinol. Invest. 29(4), 298–302 (2006). https://doi.org/10.1007/BF03344099
Y. Jing, J. Hu, R. Luo et al. Prevalence and characteristics of adrenal tumors in an unselected screening population: a cross-sectional study. Ann. Intern. Med. 175(10), 1383–1391 (2022). https://doi.org/10.7326/M22-1619
J.H. Song, F.S. Chaudhry, W.W. Mayo-Smith, The incidental adrenal mass on CT: prevalence of adrenal disease in 1,049 consecutive adrenal masses in patients with no known malignancy. AJR Am. J. Roentgenol. 190(5), 1163–1168 (2008). https://doi.org/10.2214/AJR.07.2799
N.M. Albiger, G. Occhi, F. Sanguin et al. Adrenal nodules in patients with Cushing’s disease: prevalence, clinical significance and follow-up. J. Endocrinol. Invest. 34(8), e204–e209 (2011). https://doi.org/10.3275/7349
G. Borretta, M. Terzolo, F. Cesario, I. Meineri, A. Pia, A. Angeli, Coexistence of unilateral adrenal macronodule and Cushing’s disease. Report of two cases. J. Endocrinol. Invest. 19(2), 131–135 (1996). https://doi.org/10.1007/BF03349849
M.K.M. Shakir, I.C. Ebrahim, A. Spiro, V.Q. Mai, T.D. Hoang, Coexistence of Cushing disease with a solitary adrenocorticotrophic hormone-dependent adrenal adenoma. AACE Clin. Case Rep. 8(1), 41–44 (2022). https://doi.org/10.1016/j.aace.2020.11.027
F.W.F. Hanna, S. Hancock, C. George et al. Adrenal incidentaloma: prevalence and referral patterns from routine practice in a large UK university teaching hospital. J. Endocr. Soc. 6(1), bvab180 (2022). https://doi.org/10.1210/jendso/bvab180
H.J.L.M. Timmers, E.M. van Ginneken, P. Wesseling, C.G.J. Sweep, A.R.M.M. Hermus, A patient with recurrent hypercortisolism after removal of an ACTH-secreting pituitary adenoma due to an adrenal macronodule. J. Endocrinol. Invest. 29(10), 934–939 (2006). https://doi.org/10.1007/BF03349200
J. Santos, I. Paiva, L. Gomes et al. [Recurrent hypercortisolism after removal of an ACTH secretor pituitary adenoma associated with an adrenal macronodule]. Acta Med. Port. 23(1), 107–112 (2010)
A. Tabarin, S. Magimel, F. Laurent, A. Navarranne, J. Guérin, P. Roger, [Biological and developmental aspects of macronodular adrenal hyperplasia in Cushing’s disease]. Ann. Endocrinol. 53(2), 59–66 (1992)
G. Di Dalmazi, H.J.L.M. Timmers, G. Arnaldi et al. Somatic PRKACA mutations: association with transition from pituitary-dependent to adrenal-dependent cushing syndrome. J. Clin. Endocrinol. Metab. 104(11), 5651–5657 (2019). https://doi.org/10.1210/jc.2018-02209
K. Bathon, I. Weigand, J.T. Vanselow et al. Alterations in protein kinase A substrate specificity as a potential cause of cushing syndrome. Endocrinology 160(2), 447–459 (2019). https://doi.org/10.1210/en.2018-00775
Open access funding provided by Tel Aviv University.
Author information
Authors and Affiliations
Endocrine and Diabetes Institute, Shamir Medical Center, Zerifin, Be’er Ya’akov, Israel
Efrat Markus & Shlomit Koren
Gray Faculty of Medical & Health Sciences, Tel Aviv University, Tel Aviv, Israel
Efrat Markus, Yaron Rudman, Shlomit Koren & Ilan Shimon
Institute of Endocrinology, Beilinson Hospital, Rabin Medical Center, Petah Tikva, Israel
Yaron Rudman & Ilan Shimon
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Efrat Markus, Yaron Rudman, and Ilan Shimon. The first draft of the manuscript was written by Efrat Markus and Ilan Shimon and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Cushing’s Syndrome, a rare but complex endocrine disorder characterized by excessive cortisol production, presents unique challenges and risks during pregnancy. Recent advancements in medical understanding have led to greater awareness of the implications of this syndrome when coupled with conditions like diabetes insipidus, particularly in pregnant patients. The coexistence of these disorders emphasizes the need for a multidisciplinary approach to manage these high-risk pregnancies effectively.
In a groundbreaking case report published in BMC Endocrine Disorders, researchers Hata et al. provide an illuminating examination of a pregnant patient diagnosed with Cushing’s Syndrome along with diabetes insipidus. This syndromic constellation is particularly alarming considering the metabolical and physiological adaptations that occur during pregnancy. The researchers delve deeply into the complexities presented by this rare overlap, offering insight into potential therapeutic pathways and management strategies.
Cushing’s syndrome is often the result of pituitary adenomas or adrenal tumors that result in a hypercortisolemic state. When analyzing its manifestation during pregnancy, clinicians are faced with the delicate balance of managing both maternal and fetal health. In this compelling case, the authors explore the detrimental effects of high cortisol levels and the complications that arise from diabetes insipidus on maternal health.
Diabetes insipidus in pregnancy can further complicate the management of Cushing’s syndrome. It is primarily characterized by an inability of the kidneys to concentrate urine due to a deficiency in the antidiuretic hormone (ADH). This disorder can lead to severe dehydration, electrolyte imbalances, and complications such as preterm labor or uterine atony. By detailing the clinical features of the patient, the report underscores the need for vigilant monitoring and timely interventions to prevent adverse outcomes.
Central to the case is the interplay between the hormonal milieu of pregnancy and the pathological processes of Cushing’s syndrome. The physiological increase in cortisol can mask or exacerbate the symptoms of diabetes insipidus. Thus, clinicians must be astute in recognizing the overlays of these conditions to adjust management plans accordingly. This is especially critical in the prenatal period, where traditional approaches might clash with the unique requirements of pregnancy.
Therapeutic management for such patients is multifaceted. Close collaboration among obstetricians, endocrinologists, and neonatologists is essential to ensure that both maternal and fetal welfare are prioritized. This case illustrates the complexity involved in choosing appropriate pharmacotherapy while minimizing risks to the developing fetus. Importantly, the authors suggest that non-invasive monitoring techniques may help in realizing a safer management regime.
The psychological impact on mothers grappling with these intertwined conditions cannot be overstated. The report sheds light on the emotional strain that awaits patients who must anticipate the uncertainties surrounding their pregnancies. Understanding these layers can aid healthcare providers in offering holistic support not just medically, but psychologically as well.
An often-overlooked aspect of such complex cases is the significance of postnatal follow-up. After delivery, the management of Cushing’s Syndrome may need reevaluation as hormonal levels return to baseline. In this case, the potential resolution of diabetes insipidus after childbirth rejuvenates discussions regarding long-term monitoring and treatment adherence, ensuring that mothers receive the care they need as they transition into motherhood.
Women with Cushing’s Syndrome and diabetes insipidus can experience heightened fatigue, which complicates the already demanding experience of pregnancy. The authors advocate for the integration of lifestyle modifications and supportive measures to help manage energy levels, further illustrating the multifaceted management required in such cases. These alterations can significantly contribute to improving the quality of life for these women in an already challenging scenario.
The ethical considerations surrounding the treatment of pregnant patients with rare syndromes add another layer of complexity. The authors emphasize the importance of informed consent, particularly as clinical decisions might involve experimental therapies or interventions that are not standard for pregnant patients. Open dialogues between patients and providers about risks and benefits can lead to better decision-making processes tailored to individual patient needs.
In conclusion, Hata et al.’s illuminating case report on Cushing’s Syndrome with diabetes insipidus in pregnancy serves as a pivotal reference for clinicians navigating the complexities of these coexisting conditions. As medical science continues to evolve, the insights offered in this report will undoubtedly inform best practices for managing intricate cases, further enhancing maternal-fetal medicine. The need for ongoing research and clinical trials remains crucial as we strive to optimize pregnancy outcomes in patients suffering from this rare combination of disorders.
As we look toward the future, the challenges presented by these conditions urge the medical community to prioritize collaborative care models, innovative therapeutic strategies, and comprehensive support systems for affected patients. While this case report sheds light on the clinical intricacies involved, it also heralds a call to action for further exploration into Cushing’s Syndrome and its implications in pregnancy, ensuring that mothers receive the best possible care during one of life’s most critical journeys.
Subject of Research: Cushing’s Syndrome with diabetes insipidus in pregnancy
Article Title: Cushing’s Syndrome with diabetes insipidus in pregnancy: a case report
Article References:
Hata, S., Shinokawa, N., Harada, Y. et al. Cushing’s Syndrome with diabetes insipidus in pregnancy: a case report.
BMC Endocr Disord 25, 197 (2025). https://doi.org/10.1186/s12902-025-01946-9