Cushing disease is caused by tumour in the pituitary gland which leads to excessive secretion of a hormone called adrenocorticotrophic (ACTH), which in turn leads to increasing levels of cortisol in the body. Cortisol is a steroid hormone released by the adrenal glands and helps the body to deal with injury or infection.
Increasing levels of cortisol increases the blood sugar and can even cause diabetes mellitus. However the disease is also caused due to excess production of hypothalamus corticotropin releasing hormone (CRH) which stimulates the synthesis of cortisol by the adrenal glands. The condition is named after Harvey Cushing, the doctor who first identified the disease in 1912. Cushing disease results in Cushing syndrome.
Cushing syndrome is a group of signs and symptoms developed due to prolonged exposure to cortisol. Signs and symptoms of Cushing syndrome includes hypertension, abdominal obesity, muscle weakness, headache, fragile skin, acne, thin arms and legs, red stretch marks on stomach, fluid retention or swelling, excess body and facial hair, weight gain, acne, buffalo hump, tiredness, fatigue, brittle bones, low back pain, moon shaped face etc. Symptoms vary from individual to individual depending upon the disease duration, age and gender of the patient.
Disease diagnosis is done by measuring levels of cortisol in patient’s urine, saliva or blood. For confirming the diagnosis, a blood test for ACTH is performed. The first-line treatment of the disease is through surgical resection of ACTH-secreting pituitary adenoma, however disease management is also done through medications, Cushing disease treatment market comprises of the drugs designed for lowering the level of cortisol in the body. Thus patients suffering from Cushing disease are prescribed medications such as ketoconazole, mitotane, aminoglutethimide metyrapone, mifepristone, etomidate and pasireotide.
Cushing’s disease treatment market revenue is growing with a stable growth rate, this is attributed to increasing number of pipeline drugs. Also increasing interest of pharmaceutical companies to develop Cushing disease drugs is a major factor contributing to the revenue growth of Cushing disease treatment market over the forecast period.
Current and emerging players’ focuses on physician education and awareness regarding availability of different drugs for curing Cushing disease, thus increasing the referral speeds, time to diagnosis and volume of diagnosed Cushing disease individuals.
Growing healthcare expenditure and increasing awareness regarding Cushing syndrome aids in the revenue growth of Cushing’s disease treatment market. Increasing number of new product launches also drives the market for Cushing’s disease Treatment devices. However availability of alternative therapies for curing Cushing syndrome is expected to hamper the growth of the Cushing’s disease treatment market over the forecast period.
The Cushing’s disease Treatment market is segment based on the product type, technology type and end user
Cushing’s disease Treatment market is segmented into following types:
By Drug Type
Ketoconazole
Mitotane
Aminoglutethimide
Metyrapone
Mifepristone
Etomidate
Pasireotide
By End User
Hospital Pharmacies
Retail Pharmacies
Drug Stores
Clinics
e-Commerce/Online Pharmacies
Cushing’s disease treatment market revenue is expected to grow at a good growth rate, over the forecast period. The market is anticipated to perform well in the near future due to increasing awareness regarding the condition. Also the market is anticipated to grow with a fastest CAGR over the forecast period, attributed to increasing investment in R&D and increasing number of new product launches which is estimated to drive the revenue growth of Cushing’s disease treatment market over the forecast period.
Depending on geographic region, the Cushing’s disease treatment market is segmented into five key regions: North America, Latin America, Europe, Asia Pacific (APAC) and Middle East & Africa (MEA).
North America is occupying the largest regional market share in the global Cushing’s disease treatment market owing to the presence of more number of market players, high awareness levels regarding Cushing syndrome. Healthcare expenditure and relatively larger number of R&D exercises pertaining to drug manufacturing and marketing activities in the region. Also Europe is expected to perform well in the near future due to increasing prevalence of the condition in the region.
Asia Pacific is expected to grow at the fastest CAGR because of increase in the number of people showing the symptoms of Cushing syndrome, thus boosting the market growth of Cushing’s disease treatment market throughout the forecast period.
Some players of Cushing’s disease Treatment market includes CORCEPT THERAPEUTICS, HRA Pharma, Strongbridge Biopharma plc, Novartis AG, etc. However there are numerous companies producing branded generics for Cushing disease. The companies in Cushing’s disease treatment market are increasingly engaged in strategic partnerships, collaborations and promotional activities to capture a greater pie of market share.
The research report presents a comprehensive assessment of the market and contains thoughtful insights, facts, historical data, and statistically supported and industry-validated market data. It also contains projections using a suitable set of assumptions and methodologies. The research report provides analysis and information according to categories such as market segments, geographies, types, technology and applications.
SAN DIEGO, CA, USA I August 10, 2021 I Crinetics Pharmaceuticals, Inc. (Nasdaq: CRNX), a clinical stage pharmaceutical company focused on the discovery, development, and commercialization of novel therapeutics for rare endocrine diseases and endocrine-related tumors, today announced positive preliminary findings from the single ascending dose (SAD) portion of a first-in-human Phase 1 clinical study with CRN04894 demonstrating pharmacologic proof-of-concept for this first-in-class, investigational, oral, nonpeptide adrenocorticotropic hormone (ACTH) antagonist that is being developed for the treatment of conditions of ACTH excess, including Cushing’s disease and congenital adrenal hyperplasia.
“ACTH is the central hormone of the endocrine stress response. Even though we’ve known about its clinical significance for more than 100 years, there has never been an ACTH antagonist available to intervene in diseases of excess stress hormones. This is an important milestone for the field of endocrinology and for our company,” said Scott Struthers, Ph.D., founder and chief executive officer of Crinetics. “I am extremely proud of our team that conceived, discovered and developed CRN04894 this far. This is the second molecule to emerge from our in-house discovery efforts and demonstrate pharmacologic proof of concept. I am very excited to see what it can do in upcoming clinical studies.”
The 39 healthy volunteers who enrolled in the SAD cohorts were administered oral doses of CRN04894 (10 mg to 80 mg, or placebo) two hours prior to a challenge with synthetic ACTH. Analyses of basal cortisol levels (before ACTH challenge) showed that CRN04894 produced a rapid and dose-dependent reduction of cortisol by 25-56%. After challenge with a supra-pathophysiologic dose of ACTH (250 mcg), CRN04894 suppressed cortisol (as measured by AUC) up to 41%. After challenge with a disease-relevant dose of ACTH (1 mcg), CRN04894 showed a clinically meaningful reduction in cortisol AUC of 48%. These reductions in cortisol suggest that CRN04894 is bound with high affinity to its target receptor on the adrenal gland and blocking the activity of ACTH. CRN04894 was well tolerated in the healthy volunteers who enrolled in these SAD cohorts and all adverse events were considered mild.
“We are very encouraged by these single ascending dose data which clearly demonstrate proof of ACTH antagonism with CRN04894 exposure in healthy volunteers,” stated Alan Krasner, M.D., chief medical officer of Crinetics. “We look forward to completing this study and assessing results from the multiple ascending dose cohorts. As a clinical endocrinologist, I recognize the pioneering nature of this work and eagerly look forward to further understanding the potential of CRN04894 for the treatment of diseases of ACTH excess.”
Data Review Conference Call
Crinetics will hold a conference call and live audio webcast today, August 10, 2021 at 4:30 p.m. Eastern Time to discuss the results of the CRN04894 SAD cohorts. To participate, please dial 800-772-3714 (domestic) or 212-271-4615 (international) and refer to conference ID 21996541. To access the webcast, please visit the Events page on the Crinetics website. The archived webcast will be available for 90 days.
About the CRN04894-01 Phase 1 Study
Crinetics is enrolling healthy volunteers in this double-blind, randomized, placebo-controlled Phase 1 study of CRN04894. Participants will be divided into multiple cohorts in the single ascending dose (SAD) and multiple ascending dose (MAD) phases of the study. In the SAD phase, safety and pharmacokinetics are assessed. In addition, pharmacodynamic responses are evaluated before and after challenges with injected synthetic ACTH to assess pharmacologic effects resulting from exposure to CRN04894. In the MAD phase, participants will be administered placebo or ascending doses of study drug daily for 10 days. Assessments of safety, pharmacokinetics and pharmacodynamics will also be performed after repeat dosing.
About CRN04894
Adrenocorticotropic hormone (ACTH) is synthesized and secreted by the pituitary gland and binds to melanocortin type 2 receptor (MC2R), which is selectively expressed in the adrenal gland. This interaction of ACTH with MCR2 stimulates the adrenal production of cortisol, a stress hormone that is involved in the regulation of many systems. Cortisol is involved for example in the regulation of blood sugar levels, metabolism, inflammation, blood pressure, and memory formulation, and excess adrenal androgen production can result in hirsutism, menstrual dysfunction, infertility in men and women, acne, cardiometabolic comorbidities and insulin resistance. Diseases associated with excess of ACTH, therefore, can have significant impact on physical and mental health. Crinetics’ ACTH antagonist, CRN04894, has exhibited strong binding affinity for MC2R in preclinical models and demonstrated suppression of adrenally derived glucocorticoids and androgens that are under the control of ACTH, while maintaining mineralocorticoid production.
About Cushing’s Disease and Congenital Adrenal Hyperplasia
Cushing’s disease is a rare disease with a prevalence of approximately 10,000 patients in the United States. It is more common in women, between 30 and 50 years of age. Cushing’s disease often takes many years to diagnose and may well be under-diagnosed in the general population as many of its symptoms such as lethargy, depression, obesity, hypertension, hirsutism, and menstrual irregularity can be incorrectly attributed to other more common disorders.
Congenital adrenal hyperplasia (CAH) encompasses a set of disorders that are caused by genetic mutations that result in impaired cortisol synthesis with a prevalence of approximately 27,000 patients in the United States. This lack of cortisol leads to a loss of feedback mechanisms and results in persistently high levels of ACTH, which in turn causes overstimulation of the adrenal cortex. The resulting adrenal hyperplasia and over-secretion of other steroids (particularly androgens) and steroid precursors can lead to a variety of effects from improper gonadal development to life-threatening adrenal crisis.
About Crinetics Pharmaceuticals Crinetics Pharmaceuticals is a clinical stage pharmaceutical company focused on the discovery, development, and commercialization of novel therapeutics for rare endocrine diseases and endocrine-related tumors. The company’s lead product candidate, paltusotine, is an investigational, oral, selective nonpeptide somatostatin receptor type 2 agonist for the treatment of acromegaly, an orphan disease affecting more than 26,000 people in the United States. A Phase 3 program to evaluate safety and efficacy of paltusotine for the treatment of acromegaly is underway. Crinetics also plans to advance paltusotine into a Phase 2 trial for the treatment of carcinoid syndrome associated with neuroendocrine tumors. The company is also developing CRN04777, an investigational, oral, nonpeptide somatostatin receptor type 5 (SST5) agonist for congenital hyperinsulinism, as well as CRN04894, an investigational, oral, nonpeptide ACTH antagonist for the treatment of Cushing’s disease, congenital adrenal hyperplasia, and other diseases of excess ACTH. All of the company’s drug candidates are new chemical entities resulting from in-house drug discovery efforts and are wholly owned by the company.
Bilateral adrenalectomy (BA) still plays an important role in the management of Cushing’s disease (CD). Nelson’s syndrome (NS) is a severe complication of BA, but conflicting data on its prevalence and predicting factors have been reported. The aim of this study was to determine the prevalence of NS, and identify factors associated with its development.
Data sources
Systematic literature search in four databases.
Study Selection
Observational studies reporting the prevalence of NS after BA in adult patients with CD.
Data extraction
Data extraction and risk of bias assessment were performed by three independent investigators.
Data synthesis
Thirty-six studies, with a total of 1316 CD patients treated with BA, were included for the primary outcome. Pooled prevalence of NS was 26% (95% CI 22–31%), with moderate to high heterogeneity (I2 67%, P < 0.01). The time from BA to NS varied from 2 months to 39 years. The prevalence of NS in the most recently published studies, where magnet resonance imaging was used, was 38% (95% CI 27–50%). The prevalence of treatment for NS was 21% (95% CI 18–26%). Relative risk for NS was not significantly affected by prior pituitary radiotherapy [0.9 (95% CI 0.5–1.6)] or pituitary surgery [0.6 (95% CI 0.4–1.0)].
Conclusions
Every fourth patient with CD treated with BA develops NS, and every fifth patient requires pituitary-specific treatment. The risk of NS may persist for up to four decades after BA. Life-long follow-up is essential for early detection and adequate treatment of NS.
Introduction
Cushing´s disease (CD) is a rare disorder associated with excess morbidity and increased mortality [1, 2]. Previously, bilateral adrenalectomy (BA) was the mainstay treatment for CD. During the last decades, however, other treatment modalities have emerged, including pituitary surgery, radiotherapy and medical treatments. Despite this, BA is still considered when other treatment options have failed to achieve remission, or when a rapid relief of hypercortisolism is necessary [3].
BA is considered to be a safe and effective treatment for CD [4], especially after the laparoscopic approach was introduced during the 1990s [5]. There are, however, significant drawbacks with BA, mainly the unavoidable chronic adrenal insufficiency, as well as the risk for Nelson’s syndrome (NS), i.e., growth of the remaining pituitary tumor and excessive production of ACTH, that may cause optic nerve or chiasmal compression and mucocutaneous hyperpigmentation [6].
The prevalence of NS varies between studies, mainly due to a lack of consensus on the definition and diagnostic criteria for the syndrome [7, 8]. Previously published studies are also inconsistent as to whether factors such as previous radiotherapy, age at BA, gender and duration of CD, may affect the risk of developing NS. Furthermore, high ACTH concentrations after BA have been suggested as a risk factor for developing NS [9,10,11,12].
Thus, the primary aim of this systematic review and meta-analysis was to estimate the prevalence of NS after BA for CD, both the total prevalence of NS as well the prevalence of NS requiring treatment with pituitary surgery and/or radiotherapy. The secondary aim was to investigate risk factors associated with development of NS.
Methods
A systematic review and meta-analysis was conducted according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) [13]. The PICO process was applied for the definition of the research question and eligibility criteria for the literature search. The protocol of this review was registered in the PROSPERO database (CRD42020163918).
Search strategy
We searched PubMed, Embase, Cochrane Library and Web of Science on February 25th 2020, with no start date restriction, for relevant articles by using the following terms: “Cushing´s syndrome” or “Cushing´s disease” or “Hypercortisolism” or “Pituitary ACTH hypersecretion” or “corticotroph tumor” or “corticotroph tumors” or “corticotroph adenoma” or “corticotroph adenomas” or “corticotropinoma” or “corticotropinomas” or “corticotrophinoma” or “corticotrophinomas” or “ACTH pituitary adenoma” or “ACTH pituitary adenomas” or “adrenocorticotropin pituitary adenoma” or “adrenocorticotropin pituitary adenomas” AND “bilateral adrenalectomy” or “bilateral adrenalectomies” or “total adrenalectomy” or “total adrenalectomies”. A detailed description of the search strategy is given in the Supplementary. Also, references of the included studies and relevant review articles were checked manually for additional articles. A new search was performed on January 12th 2021, prior submission, to identify any new publications.
Study selection and eligibility criteria
Eligible studies were observational studies (cohort or cross-sectional studies) reporting the prevalence of NS in adult patients with CD treated with BA. Studies including only children (age < 18 years), review articles, letters, commentaries and meeting abstracts were excluded. Moreover, case reports, case-series and studies with a population of fewer than 10 cases were excluded. Also, studies written in languages other than English were not considered for inclusion.
Data collection process and data extraction
Titles and abstracts from all identified articles were screened for eligibility and further full-text assessment by three independent investigators (EP, MP, OR). Discrepancies were resolved through discussion and consensus. Duplicate articles and studies with overlapping populations were excluded. In the latter case, the publication with the largest population, more comprehensive information on relevant clinical variables and/or lowest risk of bias was included.
Full-text assessment and data extraction were conducted independently by the same investigators as above. Data on the following predefined variables were extracted: first author, year of publication, region/hospital, study period, characteristics of the study population (number of patients, gender, follow-up, age at CD, age at BA, previous treatment with radiotherapy and/or pituitary surgery, ACTH concentrations at BA, MRI findings at CD and at BA), intervention (BA as primary or secondary treatment, remission status) and outcome (criteria for NS, number of patients with NS, age at NS, time from BA to NS, ACTH concentrations one year after BA, number of patients treated for NS, type of treatment; pituitary radiotherapy and/or pituitary surgery).
One of the studies included in the meta-analysis is our nationwide Swedish study on CD [2]. Additional clinical data, not provided in the original publication, was retrieved and used in the current analysis (Table 1).Table 1 Characteristics of the included studiesFull size table
Risk of bias assessment
The Newcastle–Ottawa Scale [14], modified to suit the current study, was used for assessment of risk of bias of all included studies. Three investigators (EP, MP, OR) assessed the studies independently, and any disagreements were resolved by discussion. Selection, comparability and outcome were assessed through predefined criteria. All studies that provided information on NS as outcome, and/or corticotroph tumor progression, were included, and the definition as well as the treatment of NS were recorded (Table 1 and Table S1). A clear definition of NS and information on treatment were considered to be two of the most important components of the quality assessment. We considered the definition of NS to be clear when it included either a new visible pituitary tumor or progression of a pituitary tumor remnant following BA, alone, or in combination with high ACTH concentrations and/or hyperpigmentation. Detailed description of the criteria for the risk of bias assessment is provided in the Supplementary file. Studies with an overall score ≥ 5 (max overall grade 8) and a clear definition of NS, were considered to have a low risk of bias.
Data synthesis and statistical analysis
Primary endpoints were the prevalence of NS, as well as the prevalence of pituitary-specific treatment for NS. Descriptive data are presented as median (range or interquartile range; IQR). Meta-analysis was performed by using the meta package in R (version 4.0.3) [15]. Statistical pooling was performed according to random-effects model due to the clinical heterogeneity among the included studies [16]. For all analyses, indices of heterogeneity, I2 statistics and Cochrane’s Q test, are reported. For the primary outcomes we estimated pooled prevalence with 95% confidence intervals (95% CI). Statistical significance was defined as P < 0.05. The possibility of publication bias was assessed by visual inspection of funnel plots as well as with the Egger’s test [17].
Sensitivity analyses were performed by excluding studies with an overall risk of bias < 5, and studies where information on diagnostic criteria for NS was lacking. By choosing the overall risk of bias < 5, all studies without adequate follow-up were also excluded (Table S2). Also, another sensitivity analysis was performed by including all studies reporting the number of patients with NS who received treatment for NS (Table 1).
Subgroup analyses were performed to investigate factors that may affect the prevalence of NS, namely pituitary radiotherapy prior to BA, prophylactic pituitary radiotherapy, overall radiotherapy (prior to BA or prophylactic), pituitary surgery (transcranial or transsphenoidal surgery) prior to BA, and BA as primary or secondary treatment. For these outcomes, we estimated relative risks (RRs), or pooled prevalence, with 95% CIs. Also, in a subgroup analysis, the prevalence (with 95% CI) of NS and treatment for NS were estimated in studies where MRI was used at diagnosis and during follow-up.
Uni- and bivariate meta-regression was used to investigate whether the prevalence of NS was influenced by median follow-up time or age at BA. The meta-analysis was performed by using the Metareg command in R. The estimated association is reported as β coefficient.
Role of funding source
The funding source had no role in the design and conduction of the study; i.e., collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Results
Identification and description of included studies
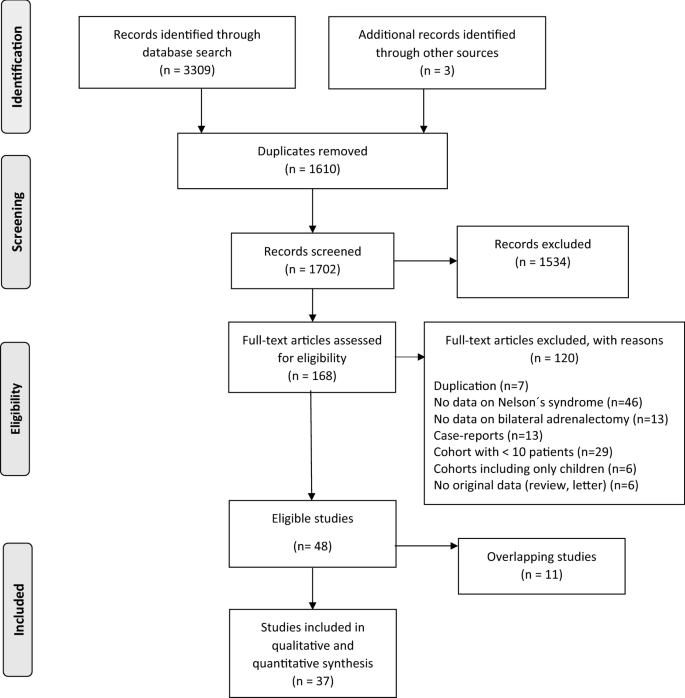
After removal of duplicates, 1702 articles were identified (Fig. 1). Three additional articles were found after checking the reference lists of identified articles and review papers. After reviewing titles, abstracts and full-text articles, 48 articles were considered eligible for further analysis. Of these, however, 11 articles were excluded due to overlapping or identical patient cohorts. Thus, 37 studies published between 1976 and 2020, were included in the current meta-analysis (Fig. 1). All studies had a retrospective observational design. Characteristics of the included studies are presented in Table 1. Two of the included studies had an overlapping cohort where one was used for the main outcome [18] and one [19] for the subgroup analyses on the influence of radiotherapy on the development of NS. An overview of risk of bias assessment of the eligible studies is provided in Table S2.
Fig. 1
In total, 1316 patients with CD treated with BA were included. The median follow-up after BA was 7 years (23 studies, range 3.3–22). Median age at BA in patients with NS was 31 years (13 studies, IQR 26–34). Median time from BA to the diagnosis of NS was 4 years (19 studies) with the shortest reported time being 2 months [20] and the longest 39 years [2]. At diagnosis of NS, hyperpigmentation was reported in 155 of 188 (82%) patients (19 studies) and chiasmal compression in 24 of 129 (19%) patients [11 studies].
Prevalence of NS
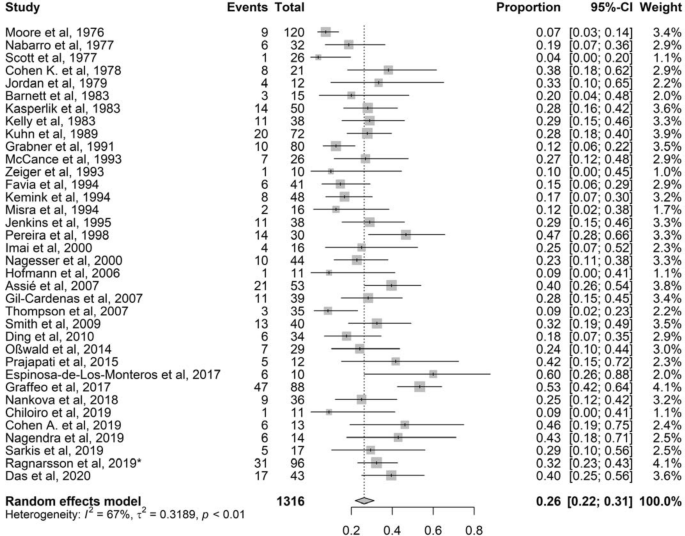
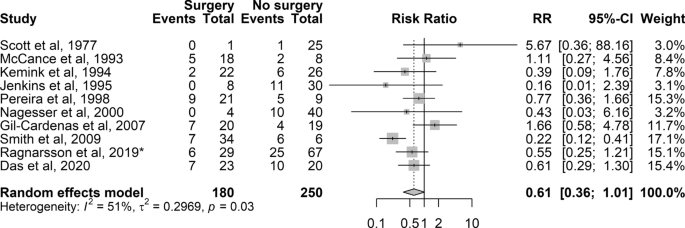
Thirty-six of 37 studies, with total 1316 patients with CD treated with BA, were included [2, 18, 20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53]. Reported prevalence of NS ranged from 4 to 60%. The mean pooled prevalence was 26% (95% CI 22–31%) with a moderate to high heterogeneity (I2 67%, P < 0.01) (Fig. 2). The Egger’s test was statistically significant (P = 0.01), but visual inspection showed no obvious asymmetry. The significant Egger’s test indicates publication bias, probably explained by the fact that case reports and cohorts with fewer than 10 participants were excluded (Fig. S1).
Fig. 2
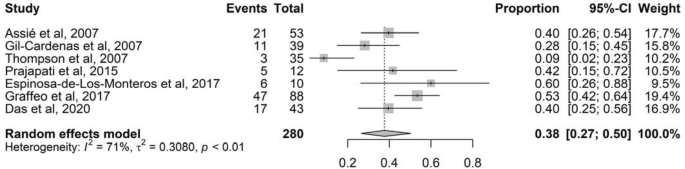
In a sensitivity analysis, excluding all studies with high risk of bias (overall score < 5) and no clear definition of NS, the pooled prevalence was 31% (95% CI 24–38%; I2 76%, 17 studies, 822 patients; P < 0.01) (Fig. S2). In a subgroup analysis, the prevalence of NS in studies where MRI was used at diagnosis and during follow-up was 38% (Fig. 3; 95% CI 27–50%; I2 71%, 7 studies, 280 patients; P < 0.01).
Fig. 3
Prevalence of treated NS
The pooled prevalence of treatment for NS was 21% (95% CI 18–26%; I2 52%, P < 0.01) (Table 1; 29 studies with 1074 patients). Thus, the pooled prevalence was slightly lower, compared to the pooled prevalence of NS in total, as well as the heterogeneity (Fig. S3). The funnel plot showed no asymmetry and Egger’s test was not statistically significant, indicating low possibility of publication bias (Fig. S4). In a subgroup analysis, the prevalence of treated NS in studies where MRI was used at diagnosis and during follow-up was 25% (95% CI 17–35%; I2 61%, 7 studies; P = 0.02).
The indication for treatment was progression of the pituitary tumor in 23 out of 28 patients (82%, five studies), optic chiasmal compression in 11 out of 91 patients (12%, 11 studies), while four patients out of 14 (one study) had both these indications for treatment. Twenty-six studies provided information on treatment modalities (pituitary surgery and/or radiotherapy). Seventy-three out of 201 patients with NS (36%) were treated with pituitary surgery, 86 (43%) with radiotherapy and 41 (20%) received both treatments.
Radiotherapy
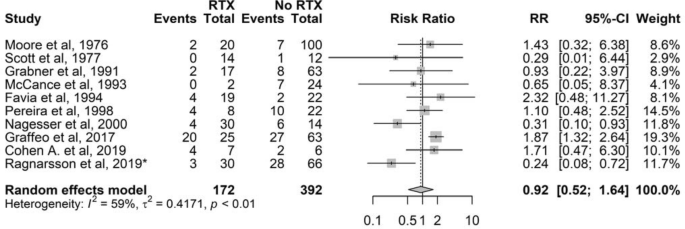
Nineteen studies provided information on radiotherapy prior to BA. However, nine studies had no events and no patients in one of the arms (radiotherapy or no radiotherapy) (Table S3). Thus, ten studies were eligible for further estimation, showing that the risk for NS in patients treated with radiotherapy prior to BA was comparable to the risk in patients not treated with radiotherapy (RR 0.9, 95% CI 0.5–1.6; 10 studies with 564 patients) (Fig. 4).
Fig. 4
Thirteen studies provided information on prophylactic radiotherapy. However, only one study provided applicable data for calculating RR, thus subgroup analysis was not performed (Table S4). In that study [20], none of the seventeen patients who received prophylactic radiotherapy developed NS, while 11 of 22 patients without radiotherapy developed NS after a mean follow-up of 4.4 years (range 10–16 years).
By using studies with information on either previous or prophylactic radiotherapy (11 studies with 603 patients; Table S5), the pooled RR was 0.8 (95% CI 0.5–1.5).
Pituitary surgery prior to BA
Of 21 studies with information on pituitary surgery prior to BA (Table S6), only ten provided information for estimation of RR. A pooled RR of 0.6 (10 studies with 430 patients; 95% CI 0.4–1.0) was found (Fig. 5), indicating that the risk for developing NS was not influenced by previous pituitary surgery.
Fig. 5
BA as primary or secondary treatment for CD
Information on whether patients with NS were treated primarily with BA or not, was provided in ten and nine studies, respectively (Fig. S5 and S6). The pooled prevalence of NS was 26% (95% CI 20–33%) for patients treated primarily with BA and 22% (95% CI 15–31%) for patients who had been treated with pituitary surgery and/or radiotherapy prior to BA.
ACTH concentrations one year after BA
Four studies provided information on ACTH concentrations during the first year after BA [45, 49, 52, 53]. In a study by Assié et al. the median ACTH concentration in patients who developed NS was 301 pmol/L, compared to 79 pmol/L in patients without NS (upper range of limit; URL 13 pmol/L) [52]. The median ACTH concentration in a study by Cohen et al. was 105 pmol/L in the NS group compared to 18 pmol/L in patients without NS (P = 0.007) (URL 10 pmol/L) [49]. Also, in a study by Das et al., there was a statistically significant difference in ACTH concentrations one year after BA between patients with and without NS (110 vs 21 pmol/L respectively; P = 0.002) [53]. On the contrary, Espinosa-de-Los-Monteros et al.found no difference in ACTH concentrations between the patients with NS and those without NS [45]. Thus, three of four studies found that high ACTH concentrations one year after BA were associated with the development of NS. However, since the ACTH assays and the conditions when ACTH was collected were different in these studies (Table S7), further comparison or a meta-analysis on ACTH levels after BA was not considered feasible.
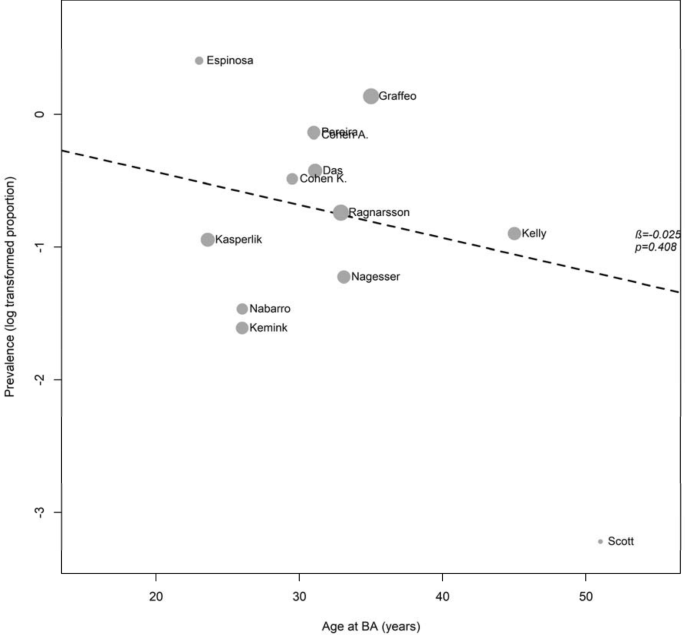
Influence of age at BA and duration of follow-up on prevalence of NS
In a meta-regression analysis, age at BA (β-coefficient = – 0.03, P = 0.4; Fig. 6) and median duration of follow-up (β-coefficient = 0.01, P = 0.7; Fig. S7) were not associated with prevalence of NS. After adjustment for follow-up, age at BA was still not associated with prevalence of NS (β-coefficient = -0.03, P = 0.4).
Fig. 6
Discussion
In this study we have for the first time evaluated the pooled prevalence of NS by using a meta-analysis on data from 36 studies, including more than 1300 patients with CD treated with BA. The overall prevalence of NS was 26% and the median time from BA to diagnosis of NS was 4 years, ranging from 0.2 to 39 years. The prevalence of patients requiring pituitary-specific treatment for NS was 21%. Furthermore, radiotherapy and pituitary surgery prior to BA, as well as age at BA, did not seem to affect the risk of developing NS.
Various definitions have been used for NS over the past decades [12]. Historically, the diagnosis was based on clinical findings related to mucocutaneous hyperpigmentation and chiasmal compression, together with signs of an enlarged sella turcica on skull radiography [6]. Since then, the diagnosis of NS in most studies has been based on (i) radiological evidence of a pituitary tumor that becomes visible, or a progression of a preexisting tumor, (ii) “high” ACTH concentrations, and (iii) hyperpigmentation [54]. In the studies with the highest prevalence of NS [45, 46], the diagnosis was based on rising ACTH concentrations and an expanding pituitary mass, where 2 mm increment in tumor size on MRI was considered to be a significant growth. On the contrary, the criteria for NS in studies with the lowest prevalence were based on hyperpigmentation, often but not always combined with a pituitary tumor responding to radiotherapy and/or a radiographic evidence of pituitary tumor on skull radiography [21, 23]. Thus, the great variance in the prevalence of NS between studies can, at least partly, be explained by the different definitions of NS. Consequently, in an expert opinion published in 2010, it was suggested that the diagnosis of NS should be based on an elevated level of ACTH >500 ng/L (110 pmol/L) in addition to rising levels of ACTH on at least three consecutive occasions and/or an expanding pituitary mass on MRI or CT following BA [54]. Similarly, in a recently published expert consensus recommendation, based on a systematic review, it was suggested that NS should be defined as radiological progression or new detection of a pituitary tumor on a thin-section MRI [55]. Furthermore, the authors recommend active surveillance with MRI three months after BA, and every 12 months for the first 3 years, and every 2–4 years thereafter, based on clinical findings. The meta-regression of the current analysis did not show an association between median follow-up time and prevalence of NS. Nevertheless, NS occurred as early as 2 months [20], and up to 39 years after BA [2], supporting that life-long surveillance after BA is necessary for patients with CD.
Active surveillance with MRI was more common in studies published during the last two decades. In fact, the use of MRI in recent studies resulted in earlier detection of a growing pituitary adenoma and, subsequently, contributed to a higher prevalence of NS. Namely, the seven studies including patients treated with BA after 1990 and using MRI reported higher prevalence of NS, both overall NS and treated NS.
Whether factors such as pituitary radiotherapy affects the risk for development of NS has been evaluated in several studies. Some studies have shown that radiotherapy prior to BA, or administrated prophylactically, can prevent or delay the development of NS [20, 39]. On the contrary, other studies have not demonstrated a protective effect of radiotherapy prior to BA [18, 37] and, moreover, one study found an association with tumor progression [46]. Nevertheless, the current meta-analysis indicates that radiotherapy prior to BA does not decrease the risk of developing NS. Neither did previous pituitary surgery affect the risk for NS.
Elevated ACTH concentrations during the first year after BA have been considered to be a strong predictor of NS [49, 52]. In fact, seven studies in the current analysis included cut-off levels for ACTH concentration, arbitrarily defined, for the diagnosis of NS [18, 25, 34, 36, 41, 45, 49]. Due to the different ACTH assays, and different conditions when ACTH was collected, no further analysis on ACTH levels was performed. Nevertheless, four studies [45, 49, 52, 53] reported ACTH concentrations one year after BA in both patients with and without NS. Three of these studies found that high ACTH concentrations one year after BA [49, 52, 53] were associated with pituitary tumor progression. Thus, these findings support the suggestion that ACTH should be monitored following BA in patients with CD [54, 55].
The prevalence of treatment for NS (21%), and the heterogeneity index (52%), were slightly lower than in the analysis of total prevalence of NS (26%, I2 67%). The majority of the patients was treated with radiotherapy, followed by pituitary surgery and combination of pituitary surgery and radiotherapy. Today, surgical removal of the pituitary tumor is considered to be the first-line therapy of NS whereas radiotherapy is considered if surgery has failed or is not possible [12, 54, 56]. In a large multi-center study by Fountas et al., the 10-year progression-free survival rates after surgery alone, or with radiotherapy, for patients with NS was 80% and 81%, respectively [57]. In comparison, progression-free survival rate in patients who did not receive treatment was 51%. Reports on the efficacy of medical therapy for NS have shown inconsistent results [56].
Strengths and limitations
This is the largest systematic review, and the first meta-analysis, on NS published to date. However, some limitations have to be acknowledged. Most important are the different diagnostic methods used to detect NS, and the different definitions of the syndrome between the studies. The majority of the studies have used the combination of hyperpigmentation, high ACTH concentrations and radiological findings for the diagnosis of NS. Notwithstanding these common criteria, there were still differences in the cut-offs of ACTH levels, the use of different radiological modalities over time as well as the radiological definition of progress of pituitary tumors. Moreover, in some studies radiological findings were used solely or in combination with either hyperpigmentation and/or bitemporal hemianopsia, ACTH concentrations or response to treatment of NS. Furthermore, in several studies a clear definition of NS was not provided. Nevertheless, we consider our attempt to address the heterogeneity of the included studies, through systematic review, quality assessment, and sensitivity and subgroup analyses to be a strength.
Conclusions
The risk of NS after BA in patients with CD is considerable and may first become clinically evident many decades later. Thus, life-long close follow-up is necessary for an early detection of a growing pituitary tumor, and adequate treatment when needed. Although this meta-analysis did not find prior surgery or radiotherapy to be associated with risk of NS, the findings are based on a limited number of studies. Thus, in order to individualize the treatment for patients with CD, further studies are needed where these and other factors possibly associated with risk of NS are evaluated.
Data availability
The data generated or analyzed during this study are included in this published article or in the Supplementary file.
Abbreviations
CD:
Cushing’s diseaseBA:
Bilateral adrenalectomyNS:
Nelson’s syndromeACTH:
Adrenocorticotropic hormoneRR:
Relative riskMRI:
Magnet resonance imagingCT:
Computer tomography
References
1.Papakokkinou E, Olsson DS, Chantzichristos D, Dahlqvist P, Segerstedt E, Olsson T, Petersson M, Berinder K, Bensing S, Hoybye C, Eden-Engstrom B, Burman P, Bonelli L, Follin C, Petranek D, Erfurth EM, Wahlberg J, Ekman B, Akerman AK, Schwarcz E, Bryngelsson IL, Johannsson G, Ragnarsson O (2020) Excess morbidity persists in patients with cushing’s disease during long-term remission: a swedish nationwide study. J Clin Endocrinol Metab 105(8):2616–2624
2.Ragnarsson O, Olsson DS, Papakokkinou E, Chantzichristos D, Dahlqvist P, Segerstedt E, Olsson T, Petersson M, Berinder K, Bensing S, Hoybye C, Eden-Engstrom B, Burman P, Bonelli L, Follin C, Petranek D, Erfurth EM, Wahlberg J, Ekman B, Akerman AK, Schwarcz E, Bryngelsson IL, Johannsson G (2019) Overall and disease-specific mortality in patients with cushing disease: a swedish nationwide study. J Clin Endocrinol Metab 104(6):2375–2384PubMedArticleGoogle Scholar
3.Nieman LK, Biller BM, Findling JW, Murad MH, Newell-Price J, Savage MO, Tabarin A, Endocrine S (2015) Treatment of cushing’s syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 100(8):2807–2831CASPubMedPubMed CentralArticleGoogle Scholar
4.Ritzel K, Beuschlein F, Mickisch A, Osswald A, Schneider HJ, Schopohl J, Reincke M (2013) Clinical review: outcome of bilateral adrenalectomy in Cushing’s syndrome: a systematic review. J Clin Endocrinol Metab 98(10):3939–3948CASPubMedArticleGoogle Scholar
5.Reincke M, Ritzel K, Osswald A, Berr C, Stalla G, Hallfeldt K, Reisch N, Schopohl J, Beuschlein F (2015) A critical reappraisal of bilateral adrenalectomy for ACTH-dependent Cushing’s syndrome. Eur J Endocrinol 173(4):M23-32CASPubMedArticleGoogle Scholar
6.Nelson DH, Meakin JW, Dealy JB Jr, Matson DD, Emerson K Jr, Thorn GW (1958) ACTH-producing tumor of the pituitary gland. N Engl J Med 259(4):161–164CASPubMedArticleGoogle Scholar
7.Guerin C, Taieb D, Treglia G, Brue T, Lacroix A, Sebag F, Castinetti F (2016) Bilateral adrenalectomy in the 21st century: when to use it for hypercortisolism? Endocr Relat Cancer 23(2):R131-142CASPubMedArticleGoogle Scholar
8.Katznelson L (2015) Bilateral adrenalectomy for Cushing’s disease. Pituitary 18(2):269–273CASPubMedArticleGoogle Scholar
9.Banasiak MJ, Malek AR (2007) Nelson syndrome: comprehensive review of pathophysiology, diagnosis, and management. Neurosurg Focus 23(3):E13PubMedArticleGoogle Scholar
10.Assie G, Bahurel H, Bertherat J, Kujas M, Legmann P, Bertagna X (2004) The Nelson’s syndrome revisited. Pituitary. 7(4):209–215PubMedArticleGoogle Scholar
11.Ragnarsson O (2020) Cushing’s syndrome disease monitoring: recurrence, surveillance with biomarkers or imaging studies. Best Pract Res Clin Endocrinol Metab. 34(2):101382PubMedArticleGoogle Scholar
12.Fountas A, Karavitaki N (2020) Nelson’s syndrome: an update. Endocrinol Metab Clin North Am 49(3):413–432PubMedArticleGoogle Scholar
13.Moher D, Liberati A, Tetzlaff J, Altman DG, Group P (2009) Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med 6(7):e1000097PubMedPubMed CentralArticleGoogle Scholar
15.Balduzzi S, Rucker G, Schwarzer G (2019) How to perform a meta-analysis with R: a practical tutorial. Evid Based Ment Health 22(4):153–160PubMedArticleGoogle Scholar
16.Lau J, Ioannidis JP, Schmid CH (1998) Summing up evidence: one answer is not always enough. Lancet 351(9096):123–127CASPubMedArticleGoogle Scholar
17.Egger M, Davey Smith G, Schneider M, Minder C (1997) Bias in meta-analysis detected by a simple, graphical test. BMJ 315(7109):629–634CASPubMedPubMed CentralArticleGoogle Scholar
18.Smith PW, Turza KC, Carter CO, Vance ML, Laws ER, Hanks JB (2009) Bilateral adrenalectomy for refractory Cushing disease: a safe and definitive therapy. J Am Coll Surg 208(6):1059–1064PubMedArticleGoogle Scholar
19.Mehta GU, Sheehan JP, Vance ML (2013) Effect of stereotactic radiosurgery before bilateral adrenalectomy for Cushing’s disease on the incidence of Nelson’s syndrome. J Neurosurg 119(6):1493–1497PubMedArticleGoogle Scholar
20.Gil-Cardenas A, Herrera MF, Diaz-Polanco A, Rios JM, Pantoja JP (2007) Nelson’s syndrome after bilateral adrenalectomy for Cushing’s disease. Surgery. 141(2):147–151
21.Moore TJ, Dluhy RG, Williams GH, Cain JP (1976) Nelson’s syndrome: frequency, prognosis, and effect of prior pituitary irradiation. Ann Intern Med 85(6):731–734CASPubMedArticleGoogle Scholar
24.Cohen KL, Noth RH, Pechinski T (1978) Incidence of pituitary tumors following adrenalectomy. A long-term follow-up study of patients treated for Cushing’s disease. Arch Internal Med 138(4):575–579CASArticleGoogle Scholar
26.Barnett AH, Livesey JH, Friday K, Donald RA, Espiner EA (1983) Comparison of preoperative and postoperative ACTH concentrations after bilateral adrenalectomy in Cushing’s disease. Clin Endocrinol (Oxf) 18(3):301–305CASArticleGoogle Scholar
28.Kelly WF, MacFarlane IA, Longson D, Davies D, Sutcliffe H (1983) Cushing’s disease treated by total adrenalectomy: long-term observations of 43 patients. Q J Med 52(206):224–231CASPubMedGoogle Scholar
29.Kuhn JM, Proeschel MF, Seurin DJ, Bertagna XY, Luton JP, Girard FL (1989) Comparative assessment of ACTH and lipotropin plasma levels in the diagnosis and follow-up of patients with Cushing’s syndrome: a study of 210 cases. Am J Med 86(6 Pt 1):678–684CASPubMedArticleGoogle Scholar
30.Grabner P, Hauerjensen M, Jervell J, Flatmark A (1991) Long-term results of treatment of cushings-disease by adrenalectomy. Acta Chirurgica- Eur J Surgery 157(8):461–464CASGoogle Scholar
31.McCance DR, Russell CF, Kennedy TL, Hadden DR, Kennedy L, Atkinson AB (1993) Bilateral adrenalectomy: low mortality and morbidity in Cushing’s disease. Clin Endocrinol 39(3):315–321CASArticleGoogle Scholar
32.Zeiger MA, Fraker DL, Pass HI, Nieman LK, Cutler GB Jr, Chrousos GP, Norton JA (1993) Effective reversibility of the signs and symptoms of hypercortisolism by bilateral adrenalectomy. Surgery 114(6):1138–1143CASPubMedGoogle Scholar
33.Favia G, Boscaro M, Lumachi F, D’Amico DF (1994) Role of bilateral adrenalectomy in Cushing’s disease. World J Surg 18(4):462–466CASPubMedArticleGoogle Scholar
34.Kemink L, Pieters G, Hermus A, Smals A, Kloppenborg P (1994) Patient’s age is a simple predictive factor for the development of Nelson’s syndrome after total adrenalectomy for Cushing’s disease. J Clin Endocrinol Metab 79(3):887–889CASPubMedGoogle Scholar
35.Misra D, Kapur MM, Gupta DK (1994) Incidence of Nelson’s syndrome and residual adrenocortical function in patients of Cushing’s disease after bilateral adrenalectomy. J Assoc Physicians India 42(4):304–305CASPubMedGoogle Scholar
36.Jenkins PJ, Trainer PJ, Plowman PN, Shand WS, Grossman AB, Wass JA, Besser GM (1995) The long-term outcome after adrenalectomy and prophylactic pituitary radiotherapy in adrenocorticotropin-dependent Cushing’s syndrome. J Clin Endocrinol Metab 80(1):165–171CASPubMedGoogle Scholar
37.Pereira MA, Halpern A, Salgado LR, Mendonca BB, Nery M, Liberman B, Streeten DH, Wajchenberg BL (1998) A study of patients with Nelson’s syndrome. Clin Endocrinol (Oxf) 49(4):533–539CASArticleGoogle Scholar
38.Imai T, Kikumori T, Funahashi H, Nakao A (2000) Surgical management of Cushing’s syndrome. Biomed Pharmacother 54(1):140–145ArticleGoogle Scholar
39.Nagesser SK, van Seters AP, Kievit J, Hermans J, Krans HM, van de Velde CJ (2000) Long-term results of total adrenalectomy for Cushing’s disease. World J Surg 24(1):108–113CASPubMedArticleGoogle Scholar
40.Hofmann BM, Fahlbusch R (2006) Treatment of Cushing’s disease: A retrospective clinical study of the latest 100 cases. Pituitary Surgery – A Modern Approach 34:158–184ArticleGoogle Scholar
41.Thompson SK, Hayman AV, Ludlam WH, Deveney CW, Loriaux DL, Sheppard BC (2007) Improved quality of life after bilateral laparoscopic adrenalectomy for Cushing’s disease: a 10-year experience. Ann Surg 245(5):790–794PubMedPubMed CentralArticleGoogle Scholar
42.Ding XF, Li HZ, Yan WG, Gao Y, Li XQ (2010) Role of adrenalectomy in recurrent Cushing’s disease. Chin Med J 123(13):1658–1662PubMedGoogle Scholar
43.Osswald A, Plomer E, Dimopoulou C, Milian M, Blaser R, Ritzel K, Mickisch A, Knerr F, Stanojevic M, Hallfeldt K, Schopohl J, Kuhn KA, Stalla G, Beuschlein F, Reincke M (2014) Favorable long-term outcomes of bilateral adrenalectomy in Cushing’s disease. Eur J Endocrinol 171(2):209–215CASPubMedArticleGoogle Scholar
44.Prajapati OP, Verma AK, Mishra A, Agarwal G, Agarwal A, Mishra SK (2015) Bilateral adrenalectomy for Cushing’s syndrome: pros and cons. Indian J Endocrinol Metabol 19(6):834–840CASArticleGoogle Scholar
45.Espinosa-de-Los-Monteros AL, Sosa-Eroza E, Espinosa E, Mendoza V, Arreola R, Mercado M (2017) Long-term outcome of the different treatment alternatives for recurrent and persistent cushing disease. Endocrine Pract: Off J Am College Endocrinol Am Assoc Clin Endocrinol 23(7):759–767ArticleGoogle Scholar
46.Graffeo CS, Perry A, Carlstrom LP, Meyer FB, Atkinson JLD, Erickson D, Nippoldt TB, Young WF, Pollock BE, Van Gompel JJ (2017) Characterizing and predicting the Nelson-Salassa syndrome. J Neurosurg 127(6):1277–1287CASPubMedArticleGoogle Scholar
47.Nankova A, Yaneva M, Elenkova A, Tcharaktchiev D, Marinov M, Hadzhiyanev A, Sechanov T, Gantchev G, Todorov G, Kirilov G, Kalinov K, Andreeva M, Zacharieva S (2018) Cushing’s syndrome: a historic review of the treatment strategies and corresponding outcomes in a single tertiary center over the past half-century. Hormone Metab Res 50(4):280–289CASArticleGoogle Scholar
48.Chiloiro S, Giampietro A, Raffaelli M, D’Amato G, Bima C, Lauretti L, Anile C, Lombardi CP, Rindi G, Bellantone R, De Marinis L, Pontecorvi A, Bianchi A (2019) Synchronous bilateral adrenalectomy in ACTH-dependent hypercortisolism: predictors, biomarkers and outcomes. Endocrine 66(3):642–649CASPubMedArticleGoogle Scholar
49.Cohen AC, Goldney DC, Danilowicz K, Manavela M, Rossi MA, Gomez RM, Cross GE, Bruno OD (2019) Long-term outcome after bilateral adrenalectomy in Cushing’s disease with focus on Nelson’s syndrome. Arch Endocrinol Metab 63(5):470–477
50.Nagendra L, Bhavani N, Pavithran PV, Kumar GP, Menon UV, Menon AS, Kumar L, Kumar H, Nair V, Abraham N, Narayanan P (2019) Outcomes of bilateral adrenalectomy in Cushing’s syndrome. Indian J Endocrinol Metab 23(2):193–197PubMedPubMed CentralArticleGoogle Scholar
51.Sarkis P, Rabilloud M, Lifante JC, Siamand A, Jouanneau E, Gay E, Chaffanjon P, Chabre O, Raverot G (2019) Bilateral adrenalectomy in Cushing’s disease: altered long-term quality of life compared to other treatment options. Ann Endocrinol 80(1):32–37ArticleGoogle Scholar
52.Assie G, Bahurel H, Coste J, Silvera S, Kujas M, Dugue MA, Karray F, Dousset B, Bertherat J, Legmann P, Bertagna X (2007) Corticotroph tumor progression after adrenalectomy in Cushing’s disease: a reappraisal of Nelson’s Syndrome. J Clin Endocrinol Metab 92(1):172–179CASPubMedArticleGoogle Scholar
53.Das L, Bhansali A, Pivonello R, Dutta P, Bhadada SK, Ahuja CK, Mavuduru R, Kumar S, Behera A, Saikia UN, Dhandapani S, Walia R (2020) ACTH increment post total bilateral adrenalectomy for Cushing’s disease: a consistent biosignature for predicting Nelson’s syndrome. Pituitary 23(5):488–497CASPubMedArticleGoogle Scholar
54.Barber TM, Adams E, Ansorge O, Byrne JV, Karavitaki N, Wass JA (2010) Nelson’s syndrome. Eur J Endocrinol 163(4):495–507CASPubMedArticleGoogle Scholar
55.Reincke M, Albani A, Assie G, Bancos I, Brue T, Buchfelder M, Chabre O, Ceccato F, Daniele A, Detomas M, Di Dalmazi G, Elenkova A, Findling J, Grossman AB, Gomez-Sanchez CE, Heaney AP, Honegger J, Karavitaki N, Lacroix A, Laws ER, Losa M, Murakami M, Newell-Price J, Pecori Giraldi F, Perez-Rivas LG, Pivonello R, Rainey WE, Sbiera S, Schopohl J, Stratakis CA, Theodoropoulou M, van Rossum EFC, Valassi E, Zacharieva S, Rubinstein G, Ritzel K (2021) Corticotroph tumor progression after bilateral adrenalectomy (Nelson’s syndrome): systematic review and expert consensus recommendations. Eur J Endocrinol. https://doi.org/10.1530/EJE-20-1088
56.Patel J, Eloy JA, Liu JK (2015) Nelson’s syndrome: a review of the clinical manifestations, pathophysiology, and treatment strategies. Neurosurg Focus 38(2):E14PubMedArticleGoogle Scholar
57.Fountas A, Lim ES, Drake WM, Powlson AS, Gurnell M, Martin NM, Seejore K, Murray RD, MacFarlane J, Ahluwalia R, Swords F, Ashraf M, Pal A, Chong Z, Freel M, Balafshan T, Purewal TS, Speak RG, Newell-Price J, Higham CE, Hussein Z, Baldeweg SE, Dales J, Reddy N, Levy MJ, Karavitaki N (2020) Outcomes of patients with Nelson’s syndrome after primary treatment: a multicenter study from 13 UK pituitary centers. J Clin Endocrinol Metab 105(5):1527–1537
We would like to thank Therese Svanberg, librarian at the Medical Library at Sahlgrenska University Hospital for her expert assistance with the literature search.
Funding
Open access funding provided by University of Gothenburg. The study was financed by grants from the Swedish state under the agreement between the Swedish government and the county councils, the ALF-agreement (ALFGBG-593301) and a grant from the Gothenburg Society of Medicine.
Author information
Affiliations
Department of Internal Medicine and Clinical Nutrition, Institute of Medicine at Sahlgrenska Academy, University of Gothenburg, 413 45, Gothenburg, SwedenEleni Papakokkinou, Marta Piasecka, Dimitrios Chantzichristos, Daniel S. Olsson, Gudmundur Johannsson & Oskar Ragnarsson
The Department of Endocrinology, Sahlgrenska University Hospital, Blå stråket 5, 413 45, Gothenburg, SwedenEleni Papakokkinou, Marta Piasecka, Dimitrios Chantzichristos, Daniel S. Olsson, Gudmundur Johannsson & Oskar Ragnarsson
Department of Environmental and Occupational Health School of Public Health and Community Medicine, University of Gothenburg, 4053, Gothenburg, SwedenHanne Krage Carlsen
Department of Public Health and Clinical Medicine, Umeå University, 901 87, Umeå, SwedenPer Dahlqvist
Department of Molecular Medicine and Surgery, Karolinska Institutet, 17176, Stockholm, SwedenMaria Petersson, Katarina Berinder, Sophie Bensing, Charlotte Höybye & Henrik Falhammar
Department of Endocrinology, Karolinska University Hospital, 171 76, Stockholm, SwedenMaria Petersson, Katarina Berinder, Sophie Bensing, Charlotte Höybye & Henrik Falhammar
Department of Endocrinology and Diabetes, Uppsala University Hospital, and Department of Medical Sciences, Endocrinology and Mineral Metabolism, Uppsala University, 751 85, Uppsala, SwedenBritt Edén Engström
Department of Endocrinology, Skåne University Hospital, University of Lund, 205 02, Malmö, SwedenPia Burman
Department of Endocrinology, Skåne University Hospital, 222 42, Lund, SwedenCecilia Follin, David Petranek & Eva Marie Erfurth
Department of Endocrinology and Department of Medical and Health Sciences, Linköping University, 581 83, Linköping, SwedenJeanette Wahlberg & Bertil Ekman
Department of Internal Medicine, School of Health and Medical Sciences, Örebro University, 702 81, Örebro, SE, SwedenJeanette Wahlberg, Anna-Karin Åkerman & Erik Schwarcz
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Papakokkinou, E., Piasecka, M., Carlsen, H.K. et al. Prevalence of Nelson’s syndrome after bilateral adrenalectomy in patients with cushing’s disease: a systematic review and meta-analysis. Pituitary (2021). https://doi.org/10.1007/s11102-021-01158-z
Background: Cushing’s syndrome is a condition caused by excessive glucocorticoid with insomnia as one of its neuropsychiatric manifestation. Cushing’s syndrome may be caused by excessive adrenocorticotropin hormone (ACTH-dependent), for example from ACTH producing pituitary tumors, or by overproduction of cortisol by adrenocortical tumors. In this report, we presented a case with Cushing’s syndrome manifesting as chronic insomnia with adrenal cortical adenoma and pituitary microadenoma.
Case presentation: A 30-year-old woman was consulted from the Neurologic Department to the Internal Medicine Department with the chief complaint of insomnia and worsening headache for 6 months prior to the admission. She had undergone head MRI and abdominal CT scan previously and was found to have both pituitary microadenoma and left adrenal mass. From the physical examination she had clinical signs of Cushing’s syndrome like Cushingoid face and purplish striae on her stomach. Midnight cortisol serum examination was done initially and showed high level of cortisol. High dose dexamethasone suppression test or DST (8 mg overnight) was later performed to help determine the main cause of Cushing’s syndrome. The result failed to reach 50% suppression of cortisol serum, suggestive that the Cushing’s syndrome was not ACTH-dependent from the pituitary but potentially from overproduction of cortisol by the left adrenal mass. Therefore, left adrenalectomy was performed and the histopathological study supported the diagnosis of adrenal cortical adenoma.
Conclusion: Chronic insomnia is a very important symptoms of Cushing’s syndrome that should not be neglected. The patient had both microadenoma pituitary and left adrenal mass thus high dose DST test (8 mg overnight) needed to be performed to differentiate the source of Cushing’s syndrome. The result showed only little suppression therefore the pituitary microadenoma was not the source of Cushing’s syndrome and more suggestive from the adrenal etiology.
Please note that if you buy through links in this article, Medical News Today may earn a small commission. Here’s their process.
Cortisol is a hormone with various functions throughout the body. However, if a person’s body cannot regulate their cortisol levels, it could lead to a serious health condition. In these cases, home cortisol tests may be useful to indicate when someone might need medical attention.
All these functions make cortisol a vital part of maintaining overall health. If the body can no longer regulate cortisol levels, it can lead to several health disorders, such as Cushing’s syndrome and Addison’s disease. Without treatment, these conditions could cause life threatening complications.
The body requires certain cortisol levels during times of stress, such as:
There are several home cortisol tests available to purchase over the counter or online. These allow a person to take a sample of blood, urine, or saliva before sending it off for analysis.
After taking a home cortisol test, people can usually receive their results within 2–5 days online or via a telephone call with a healthcare professional.
However, there are currently no studies investigating the reliability of these home cortisol tests. Therefore, people should follow up on their test results with a healthcare professional.
A test can help individuals check their cortisol levels. If the test results show these levels are too high or too low, people should seek medical advice.
A cortisol imbalance may be a sign of an underlying condition, which can lead to serious complications without treatment.
If a person cannot carry out a home cortisol test, they should speak to a medical professional who can arrange a cortisol test at a healthcare facility.
At a clinic or hospital setting, a medical professional will usually take a blood sample and analyze it for an individual’s cortisol levels.
Home cortisol tests involve a person taking a sample of blood, urine, or saliva. There are currently no studies investigating the accuracy of these results.
However, home cortisol tests may be faster and more convenient than making an appointment with a doctor to take a sample.
People may consider several factors when deciding to purchase a home cortisol test, including:
Sample type: Some tests require a blood sample, while others need a sample of urine or saliva. With this in mind, a person may wish to buy a product that uses a testing method they are comfortable providing.
Test analysis: A person may wish to purchase a product from a company that sends tests to Clinical Laboratory Improvement Amendments (CLIA)-certified labs for analysis. The Food and Drug Administration (FDA), Center for Medicaid Services, and the Centers for Disease Control and Prevention (CDC) regulate these labs to help ensure safety and accuracy.
Accuracy: Individuals may wish to speak to a pharmacist or other healthcare professional before purchasing to ensure the test is reliable and accurate.
This cortisol test uses the finger prick method to draw blood for the sample.
Here are the steps to take and send off a blood sample:
Individuals fill in their details on the collection box and activate their testing kit online at the LetsGetChecked website.
People need to wash their hands with warm soapy water before using an alcohol swab to clean the finger that they will prick.
Once the finger is completely dry, individuals pierce the skin using the lancet in the test kit. A person must wipe away the first drop of blood before squeezing some into the blood collection tube.
After closing the tube, individuals must invert it 5–10 times before placing it in the included biohazard bag, which they then place in the box.
After following these steps, people can send the sample back to LetsGetChecked using the kit’s prepaid envelope. Test results usually come back within 2–5 days.
LetsGetChecked tests samples in the same labs that primary care providers, hospitals, and government schemes use. These labs are CLIA-certified and CAP-accredited.
The company also has a team of nurses and doctors available 24 hours a day, 7 days a week, to offer ongoing support. These healthcare professionals are on hand to discuss a person’s results with them over the phone.
This Everlywell product uses a urine sample to test a person’s cortisol levels.
The test measures the levels of three hormones in a person’s body: cortisol, cortisone, and melatonin. It also measures a person’s creatinine levels.
There are three steps with this test:
Individuals register their testing kit on Everlywell’s website.
A person follows the instructions carefully to take their urine sample.
Once they have their urine sample, they place it in the prepaid package and send it off to Everlywell’s labs.
Within a few days, individuals will receive their results digitally via the Everlywell website. Medical professionals can also offer helpful insights via their secure platform.
As well as sending a personalized report of each marker, Everlywell also sends detailed information about what the results mean.
The labs where Everlywell tests samples all carry certification with CLIA. The company also ensures that all results are reviewed and certified by independent board-certified physicians within the person’s specific state.SHOP NOW
Healthlabs offers a cortisol test that tests a person’s cortisol levels twice — once in the morning and once in the evening.
The company says they do this because a person’s cortisol levels fluctuate throughout the day. Therefore, by testing twice, they can gather information on this fluctuation.
This test uses a blood sample, which a person takes once in the morning and once in the afternoon. They must follow the instructions clearly to ensure they take suitable samples.
The manufacturer says that people should collect a morning sample between 7–9 a.m. and an evening sample between 3–5 p.m.
They then need to send off their sample for analysis. After testing is complete at a CLIA-certified lab, a person will receive their results, which usually takes between 1–2 days. SHOP NOW
A person should undergo a cortisol test if they believe they may have high or low cortisol levels.
They can do this at home or speak with a medical professional who can carry out the test for them.
People may also wish to seek medical help if they show signs of too much or too little cortisol. This could indicate a potentially serious underlying health issue.
Cortisol is an important hormone that affects almost all parts of the body. It has many functions, including reducing inflammation, regulating metabolism, and controlling blood pressure.
If a person believes they have high or low cortisol levels, they may wish to take a cortisol test. Usually, these tests take place at a medical practice. However, several home cortisol tests are available to purchase.
A person can take these tests at home by providing a urine, blood, or saliva sample. Once a lab analyzes the test, people usually receive their results within a few days. Individuals should follow up any test results with a healthcare professional. No clinics, no stress. Test your cortisol levels from home
Test your cortisol level from home with LetsGetChecked. Get free shipping, medical support, and results from accredited labs within 2–5 days. Order today for 30% off. LEARN MORE