


Highlights
•
The most common cause of ectopic ACTH syndrome is pulmonary carcinoid tumors and squamous cell lung cancer; however it is a relatively uncommon complication of pulmonary neoplasms.
•
The most common cause of Cushing syndrome is iatrogenic corticosteroid use and it should be considered in all patients regardless of clinical background.
•
Low urine cortisol levels may be associated with exogenous glucocorticoid exposure.
•
Occult glucocorticoid exposure is rare but can be evaluated with liquid chromatography.
•
Consumers should be aware of the potential risks of taking supplements, especially those advertised as joint pain relief products.
Abstract
Background
Well differentiated bronchial neuroendocrine neoplasms often follow a clinically indolent course and rarely cause Ectopic ACTH syndrome. Iatrogenic corticosteroid use is the most common cause of Cushing syndrome and should be considered in all patients regardless of clinical background.
Case report
A 59 year old woman with an 11 year history of a 1.5 cm well differentiated bronchial carcinoid, presented with Cushingoid features. Laboratory results were not consistent with an ACTH dependent Cushing Syndrome and exogenous steroids were suspected. The patient received an FDA alert regarding a glucosamine supplement she had started 4 months prior for joint pain.
Discussion
Ectopic ACTH production is reported in less than 5% of patients with squamous cell lung cancer and 3% of patients with lung or pancreatic (non-MEN1) neuroendocrine tumors. Factitious corticoid exposure is rare and can be evaluated with synthetic corticosteroid serum testing.
Conclusion
Cushing syndrome due to supplements containing unreported corticosteroid doses should be considered in patients with typical Cushingoid features and contradictory hormonal testing.
1. Introduction
Well differentiated bronchial neuroendocrine neoplasms often follow a clinically indolent course and can rarely exhibit Cushing syndrome due to ectopic production of adrenocorticotropic hormone (ACTH). However the most common cause of Cushing syndrome is iatrogenic corticosteroid use and should be considered in all patients regardless of clinical background (see Fig. 1, Fig. 2, Fig. 3, Fig. 4).

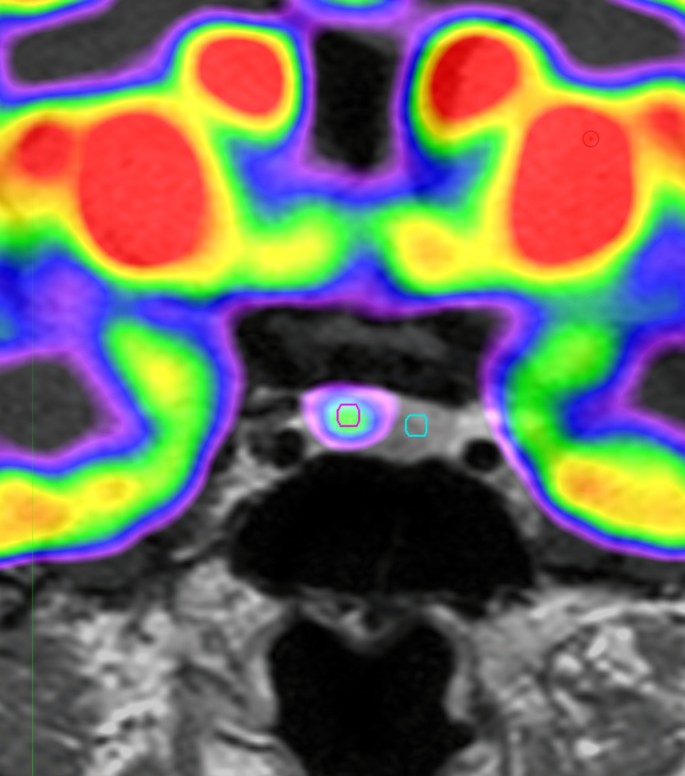
Fig. 1. DOTATATE PET/CT demonstrates a right upper lobe pulmonary nodule with intense uptake.

Fig. 2. DOTATATE PET/CT demonstrates intense uptake within a right upper lobe pulmonary nodule, consistent with biopsy-proven carcinoid tumor. There are no distant sites of abnormal uptake to suggest metastatic disease.

Fig. 3. Artri Ajo King Supplement (Source: FDA). The label claims that the product contains glucosamine, chondroitin, collagen, vitamin C, curcumin, nettle, omega 3, and methylsulfonylmethane.

Fig. 4. Artri King Supplement (Source: FDA).
2. Case report
A 59–year old woman with an 11 year history of a 1.5 cm well-differentiated bronchial carcinoid, presented with 20 lb. weight gain, facial swelling, flushing, lower extremity edema and shortness of breath over 3 months. On exam, the patient was normotensive, centrally obese with mild hirsutism, facial fullness and ruddiness with evidence of a dorsocervical fat pad. Initially there was concern for hormonal activation of her known bronchial carcinoid. Testing resulted in a normal 24-hour urine 5-HIAA (6 mg/d, n < 15 mg/dL), elevated chromogranin A (201 ng/mL, n < 103 ng/mL), normal histamine (<1.5 ng/mL, n < 1.7 ng mL), low-normal 7 AM serum cortisol (5.1 μg/dL, n 3.6–19.3 μg/dL), normal 7 AM ACTH (17 pg/mL, n < 46 pg/mL) and a surprisingly low 24-hr urinary free cortisol (1.8 mcg/hr, n 4.0–50.0 mcg/hr). A late night saliva cortisol was 0.03 mcg/dL (n 3.4–16.8 mcg/dL). Testosterone, IGF-1, glucose and electrolytes were appropriate. An echocardiogram showed an ejection fraction of 60% with no evidence of carcinoid heart disease. A Dotatate PET-CT was obtained to evaluate for progression of the neuro-endocrine tumor and revealed a stable right upper lobe pulmonary nodule with no evidence of metastatic disease. Given low cortisol levels, ectopic Cushing syndrome was excluded and exogenous steroids were suspected, however the patient denied use of oral,inhaled, or injected steroids. A cosyntropin stimulation study yielded a pre-stimulation cortisol 6.2 μg/dL with an adequate post-stimulation cortisol 23.5 μg/dL. At this stage of evaluation, the patient received an FDA alert regarding a glucosamine supplement she had started 4 months prior for joint pain. The notification advised of hidden drug ingredients including dexamethasone, diclofenac, and methocarbamol contained within Artri King Glucosamine supplements not listed on the product label but verified by FDA lab analysis. The FDA had received several adverse event reports including liver toxicity and even death associated with such products. The patient’s symptoms gradually improved after discontinuation of the supplement.
3. Discussion
3.1. Ectopic ACTH syndrome
This patient’s Cushingoid features were initially suspected to be secondary to the known bronchial neuroendocrine tumor. Ectopic ACTH production accounts for about 5–10% of all Cushing Syndrome cases [1]. The most common location of ectopic ACTH is the lungs with pulmonary carcinoid tumors being the most common cause, followed by squamous cell lung cancer [2]. Despite this patient’s history of bronchial carcinoid tumor and positive chromogranin histopathological marker, her laboratory results were not consistent with an ACTH dependent Cushing Syndrome. In fact, Cushing syndrome is a relatively uncommon neuroendocrine neoplasm complication. The prevalence of ectopic ACTH production in patients with lung tumors is rare, at less than 5% in squamous cell lung cancer and about 3% in patients with lung or pancreatic (non-MEN1) neuroendocrine tumors1.
Patients with ACTH dependent Cushing syndrome not suspected to originate from the pituitary, undergo further testing to evaluate for an ectopic ACTH secreting tumor. These tests include conventional imaging of the chest, abdomen and pelvis, as well as functional imaging such as octreotide scans, fluoride 18-fluorodeoxyglucose-positron emission tomography [18F-FDG PET], and gallium-68 DOTATATE positron emission tomography-computed tomography [Dotatate PET-CT] scan [3]. In our literature review, we found that there was insufficient evidence to determine the sensitivity and specificity of nuclear medicine imaging techniques [4,5]. In this case, the patient had no laboratory evidence for ACTH dependent Cushing Syndrome, but given the known bronchial carcinoid tumor, a repeat Dotatate PET-CT scan was obtained which demonstrated no indication of growth or spread of the known bronchial tumor.
3.2. Supplement induced Cushing Syndrome
One of the most remarkable findings in this case was the patient’s low urine cortisol level in the setting of her overt Cushingoid features. In our survey of the literature, we found that low urine cortisol levels were associated with exogenous glucocorticoid use [6,7]. The low urine cortisol levels may be reflective of intermittent glucocorticoid exposure. Indeed, this patient’s Cushingoid features were determined to be secondary to prolonged use of Artri King supplement.
Occult glucocorticoid use is difficult to diagnose even after performing a thorough medication reconciliation as patients may unknowingly consume unregulated doses of glucocorticoids in seemingly harmless supplements and medications. The incidence of supplement induced Cushing Syndrome is currently unknown as supplements are not regularly tested to detect hidden glucocorticoid doses. Additionally, the likelihood of developing supplement induced Cushing syndrome is dependent on dosage and duration of use.
In our literature review we found nine published articles describing supplement induced Cushing Syndrome [[7], [8], [9], [10], [11], [12], [13], [14], [15]], one case report of tainted counterfeit medication causing Cushing Syndrome [16], and two cases of substances with probable glucocorticoid-like activity [17,18]. Of the nine published articles of supplement induced Cushing Syndrome, six were associated with supplements marketed as arthritic joint pain relief products including ArtriKing, Maajun, and AtriVid [[7], [8], [9], [10], [11], [12]]. These products later received government issued warnings in Mexico, Malaysia, and Colombia respectively [[19], [20], [21]].
To our knowledge there have been four published reports of ArtiKing supplement induced Cushing Syndrome [[7], [8], [9], [10]]. The first documented cases were reported in 2021 in Vera Cruz, Mexico; since then the Mexican medical community reported seeing a disproportionate increase in cases of iatrogenic Cushing Syndrome due to these supplements [7]. There have also been three American published articles describing a total of 4 cases of ArtriKing supplement induced Cushing syndrome [[8], [9], [10]]. In January 2022 the FDA issued a warning about Atri Ajo King containing diclofenac, which was not listed in the product label [22]. In April 2022 the FDA expanded its warning, advising consumers to avoid all Artri and Ortiga products after the FDA found these products contained dexamethasone and diclofenac [23]. In October 2022 the FDA issued warning letters to Amazon, Walmart, and Latin Foods market for distributing Artri and Ortiga products [24].
Many supplements are not regulated by the government and may contain hidden ingredients such as glucocorticoids. In these cases further evaluation of suspected products [25], medications [16], and patient serum [26] and urine [6] utilizing techniques such as liquid chromatography may be used to confirm occult glucocorticoid exposure.
This case highlights the importance of educating patients to exercise caution when purchasing health products both online and abroad. Consumers should be aware of the potential risks of taking supplements, especially those advertised as joint pain relief products.
4. Conclusion
Although the most common cause of ectopic ACTH syndrome is pulmonary carcinoid tumors and squamous cell lung cancer, it is a relatively uncommon complication of pulmonary neoplasms.
Exogenous Cushing syndrome due to supplements containing unreported corticosteroid doses should be considered in patients with typical Cushingoid features and contradictory hormonal testing. Occult glucocorticoid exposure is rare but can be evaluated with liquid chromatography. This case report emphasizes the importance of teaching patients to be vigilant and appropriately research their health supplements.
Patient consent
Formal informed consent was obtained from the patient for publication of this case report.
Declaration of competing interest
The authors (Tomas Morales and Shanika Samarasinghe) of this case report declare that they have no financial conflicts of interest. Shanika Samrasinghe is an editorial member of the Journal of Clinical and Translational Endocrinology: Case Reports, and declares that she was not involved in the peer review and editorial decision making process for the publishing of this article.
References
- [1]
A.R. Hayes, A.B. GrossmanThe ectopic adrenocorticotropic hormone syndrome: rarely easy, always challengingEndocrinol Metab Clin N Am, 47 (2) (2018 Jun), pp. 409-425, 10.1016/j.ecl.2018.01.005PMID: 29754641
- [2]
A.M. Isidori, A. LenziEctopic ACTH syndromeArq Bras Endocrinol Metabol, 51 (8) (2007 Nov), pp. 1217-1225, 10.1590/s0004-27302007000800007PMID: 18209859
- [3]
J. Young, M. Haissaguerre, O. Viera-Pinto, O. Chabre, E. Baudin, A. TabarinManagement of endocrine disease: cushing’s syndrome due to ectopic ACTH secretion: an expert operational opinionEur J Endocrinol, 182 (4) (2020 Apr), pp. R29-R58, 10.1530/EJE-19-0877PMID: 31999619
- [4]
E. Varlamov, J.M. Hinojosa-Amaya, M. Stack, M. FleseriuDiagnostic utility of Gallium-68-somatostatin receptor PET/CT in ectopic ACTH-secreting tumors: a systematic literature review and single-center clinical experiencePituitary, 22 (5) (2019 Oct), pp. 445-455, 10.1007/s11102-019-00972-wPMID: 31236798
- [5]
A.M. Isidori, E. Sbardella, M.C. Zatelli, M. Boschetti, G. Vitale, A. Colao, R. Pivonello, ABC Study GroupConventional and nuclear medicine imaging in ectopic cushing’s syndrome: a systematic reviewJ Clin Endocrinol Metab, 100 (9) (2015 Sep), pp. 3231-3244, 10.1210/JC.2015-1589PMID: 26158607; PMCID: PMC4570166
- [6]
G. Cizza, L.K. Nieman, J.L. Doppman, M.D. Passaro, F.S. Czerwiec, G.P. Chrousos, G.B. Cutler Jr.Factitious cushing syndromeJ Clin Endocrinol Metab, 81 (10) (1996 Oct), pp. 3573-3577, 10.1210/jcem.81.10.8855803PMID: 8855803
- [7]
R. Patel, S. Sherf, N.B. Lai, R. YuExogenous cushing syndrome caused by a “herbal” supplementAACE Clin Case Rep, 8 (6) (2022 Aug 5), pp. 239-242, 10.1016/j.aace.2022.08.001PMID: 36447831; PMCID: PMC9701910
- [8]
C. Dunn, J. Amaya, P. GreenA case of iatrogenic cushing’s syndrome following use of an over-the-counter arthritis supplement2023Case Rep Endocrinol (2023 Mar 11), Article 4769258, 10.1155/2023/4769258PMID: 36941974; PMCID: PMC10024620
- [9]
N. Mikhail, K. Kurator, E. Martey, A. Gaitonde, C. Cabrera, P. BalingitIatrogenic cushing’s syndrome caused by adulteration of a health product with dexamethasoneInt J Endovascul Treatment Innovat Techn, 3 (1) (2022 Nov 23), pp. 6-9
- [10]
L. Del Carpio-Orantes, A.Q. Barrat-Hernández, A. Salas-GonzálezIatrogenic Cushing syndrome due to fallacious herbal supplements. The case of ortiga ajo rey and Artri kingColegio de Medicina Interna de México, 37 (4) (2021), pp. 599-602
- [11]
F. Wahab, R.A. Rahman, L.H. Yaacob, N.M. Noor, N. DramanA case report of steroid withdrawal syndromeKorean J Fam Med, 41 (5) (2020 Sep), pp. 359-362, 10.4082/kjfm.18.0181Epub 2020 Sep 18. PMID: 32961047; PMCID: PMC7509117
- [12]
M. Zuluaga Quintero, A. Ramírez, A. Palacio, J.F. Botero, A. ClavijoSíndrome de Cushing exógeno e insuficiencia adrenal relacionada con consumo de producto naturalActa Méd Colomb, 42 (4) (2017), pp. 243-246, 10.36104/amc.2017.1006
- [13]
R. Patell, R. Dosi, S. Sheth, P. JariwalaAverting a crisis by ‘add’ing up the clues2014:bcr2014204685BMJ Case Rep (2014 Jun 2), 10.1136/bcr-2014-204685PMID: 24891489; PMCID: PMC4054156
- [14]
H. HendartoIatrogenic Cushing’s syndrome caused by treatment with traditional herbal medicine, a case report1st International Integrative Conference on Health, Life and Social Sciences (ICHLaS 2017) (2017 Dec), 10.2991/ichlas-17.2017.9Atlantis Press
- [15]
P.C. Oldenburg-Ligtenberg, M.M. van der WesterlakenA woman with Cushing’s syndrome after use of an Indonesian herb: a case reportNeth J Med, 65 (4) (2007 Apr), pp. 150-152PMID: 17452765
- [16]
F. Azizi, A. Jahed, M. Hedayati, M. Lankarani, H.S. Bejestani, F. Esfahanian, N. Beyraghi, A. Noroozi, F. KobarfardOutbreak of exogenous Cushing’s syndrome due to unlicensed medicationsClin Endocrinol, 69 (6) (2008 Dec), pp. 921-925, 10.1111/j.1365-2265.2008.03290.xEpub 2008 May 6. PMID: 18462262
- [17]
C. Martini, E. Zanchetta, M. Di Ruvo, A. Nalesso, M. Battocchio, E. Gentilin, E. Degli Uberti, R. Vettor, M.C. ZatelliCushing in a leaf: endocrine disruption from a natural remedyJ Clin Endocrinol Metab, 101 (8) (2016 Aug), pp. 3054-3060, 10.1210/jc.2016-1490Epub 2016 May 24. PMID: 27218272
- [18]
A.J. Razenberg, J.W. Elte, A.P. Rietveld, H.C. van Zaanen, M.C. CabezasA ‘smart’ type of Cushing’s syndromeEur J Endocrinol, 157 (6) (2007 Dec), pp. 779-781, 10.1530/EJE-07-0538PMID: 18057386
- [19]
COFEPRIS (Federal Committee for Protection from Sanitary Risks)Public notification: COFEPRIS alerts about the illegal marketing of the product “ARTRI AJO KING”, Which does not have a sanitary registration
- [20]
Ministry of Health MalaysiaPublic notification: the truth about Maahun/Jamu
- [21]
INVIMA (National Food and Drug Surveillance Institute of Colombia)Health Alert: safety information about the product “ARTRIVID PLUS” promoted in different media of the country
- [22]
FDAPublic notification: Artri ajo king contains hidden drug ingredient
- [23]
FDAPublic Notification: Artri King contains hidden drug ingredients
- [24]
FDA warns consumers not to purchase or use Artri and Ortiga products, which may contain hidden drug ingredients
- [25]
P. Kempegowda, L. Quinn, L. Shepherd, S. Kauser, B. Johnson, A. Lawson, A. BatesAdrenal insufficiency from steroid-containing complementary therapy: importance of detailed historyEndocrinol Diabetes Metab Case Rep, 2019 (1) (2019 Jul 26), pp. 1-4, 10.1530/EDM-19-0047PMID: 31352697; PMCID: PMC6685090
- [26]
M.M. Pineyro, L. Redes, S. De Mattos, L. Sanchez, E. Brignardello, V. Bianchi, V. Ems, D. Centurión, M. ViolaFactitious cushing’s syndrome: a diagnosis to consider when evaluating hypercortisolismFront Endocrinol, 10 (2019 Mar 4), p. 129, 10.3389/fendo.2019.00129PMID: 30886602; PMCID: PMC6409302
Filed under: Cancer, Cushing's, Rare Diseases | Tagged: ACTH, Cushing's Syndrome, ectopic, iatrogenic, Lung carcinoid, squamous cell lung cancer, supplement, urine | Leave a comment »


 View Full Size
View Full Size View Full Size
View Full Size View Full Size
View Full Size View Full Size
View Full Size View Full Size
View Full Size