


Medications, Surgery, and Other Treatments for Cushing’s Syndrome
Written by Julie M. Gentile | Reviewed by Daniel J. Toft MD, PhD
Treatment for Cushing’s syndrome depends on what symptoms you’re experiencing as well as the cause of Cushing’s syndrome.
Cushing’s syndrome is caused by an over-exposure to the hormone cortisol. This excessive hormone exposure can come from a tumor that’s over-producing either cortisol or adrenocorticotropic hormone (ACTH—which stimulates the body to make cortisol). It can also come from taking too many corticosteroid medications over a long period of time; corticosteroids mimic the effect of cortisol in the body.
The goal of treatment is to address the over-exposure. This article walks you through the most common treatments for Cushing’s syndrome.
Gradually decreasing corticosteroid medications: If your doctor has identified that the cause of your Cushing’s syndrome is corticosteroid medications, you may be able to manage your Cushing’s syndrome symptoms by reducing the overall amount of corticosteroids you take.
It’s common for some people with certain health conditions—such as arthritis and asthma—to take corticosteroids to help them manage their symptoms. In these cases, your doctor can prescribe non-corticosteroid medications, which will allow you to reduce—or eliminate—your use of corticosteroids.
It’s important to note that you shouldn’t stop taking corticosteroid medications on your own—suddenly stopping these medications could lead to a drop in cortisol levels—and you need a healthy amount of cortisol. When cortisol levels get too low, it can cause a variety of symptoms, such as muscle weakness, fatigue, weight loss, and low blood pressure, which may be life-threatening.
Instead, your doctor will gradually reduce your dose of corticosteroids to allow your body to resume normal production of cortisol.
If for some reason you cannot stop taking corticosteroids, your doctor will monitor your condition very carefully, frequently checking to make sure your blood glucose levels as well as your bone mass levels are normal. Elevated blood glucose levels and low bone density are signs of Cushing’s syndrome.
Surgery to remove a tumor: If it’s a tumor causing Cushing’s syndrome, your doctor may recommend surgery to remove the tumor. The 2 types of tumors that can cause Cushing’s are pituitary tumors (also called pituitary adenomas) and adrenal tumors. However, other tumors in the body (eg, in the lungs or pancreas) can cause Cushing’s syndrome, too.
Pituitary adenomas are benign (non-cancerous), and most adrenal tumors are as well. However, in rare cases, adrenal tumors can be malignant (cancerous). These tumors are called adrenocortical carcinomas, and it’s important to treat them right away.
Surgery for removing a pituitary tumor is a delicate process. It’s typically performed through the nostril, and your surgeon will use tiny specialized tools. The success, or cure, rate of this procedure is more than 80% when performed by a surgeon with extensive experience. If surgery fails or only produces a temporary cure, surgery can be repeated, often with good results.
If you have surgery to remove an adrenal tumor or tumor in your lungs or pancreas, your surgeon will typically remove it through a standard open surgery (through an incision in your stomach or back) or minimally invasive surgery in which small incisions are made and tiny tools are used.
In some cases of adrenal tumors, surgical removal of the adrenal glands may be necessary.
Radiation therapy for tumors: Sometimes your surgeon can’t remove the entire tumor. If that happens, he or she may recommend radiation therapy—a type of treatment that uses high-energy radiation to shrink tumors and/or destroy cancer cells.
Radiation therapy may also be prescribed if you’re not a candidate for surgery due to various reasons, such as location or size of the tumor. Radiation therapy for Cushing’s syndrome is typically given in small doses over a period of 6 weeks or by a technique called stereotactic radiosurgery or gamma-knife radiation.
Stereotactic radiosurgery is a more precise form of radiation. It targets the tumor without damaging healthy tissue.
With gamma-knife radiation, a large dose of radiation is sent to the tumor, and radiation exposure to the healthy surrounding tissues is minimized. Usually one treatment is needed with this type of radiation.
Medications for Cushing’s syndrome: If surgery and/or radiation aren’t effective, medications can be used to regulate cortisol production in the body. However, for people who have severe Cushing’s syndrome symptoms, sometimes medications are used before surgery and radiation treatment. This can help control excessive cortisol production and reduce risks during surgery.
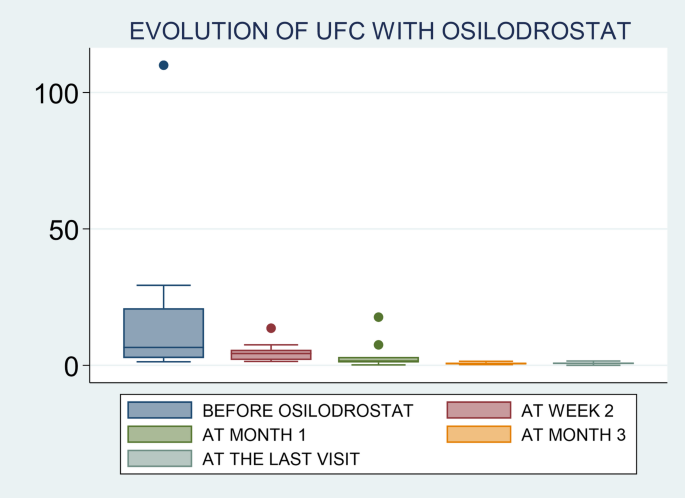
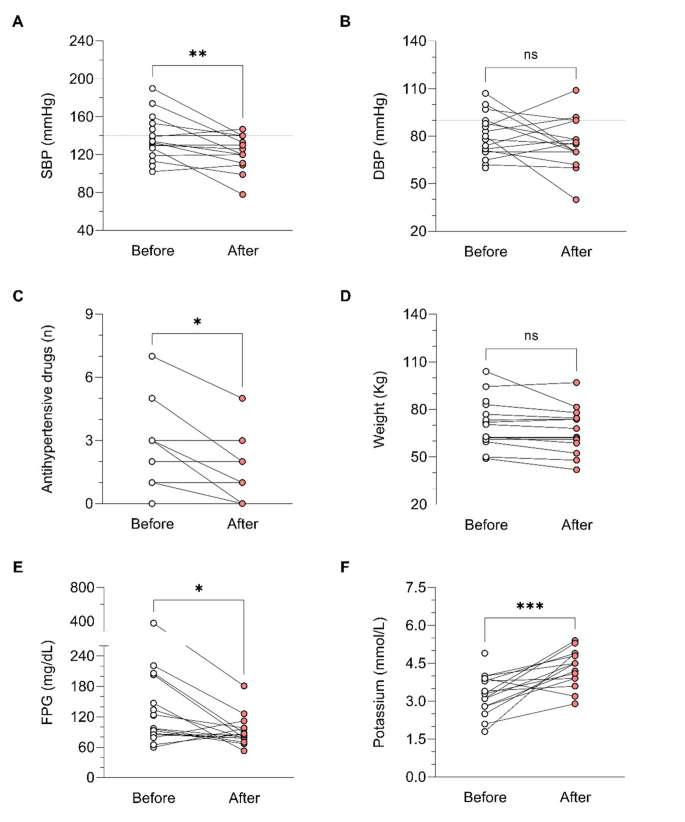
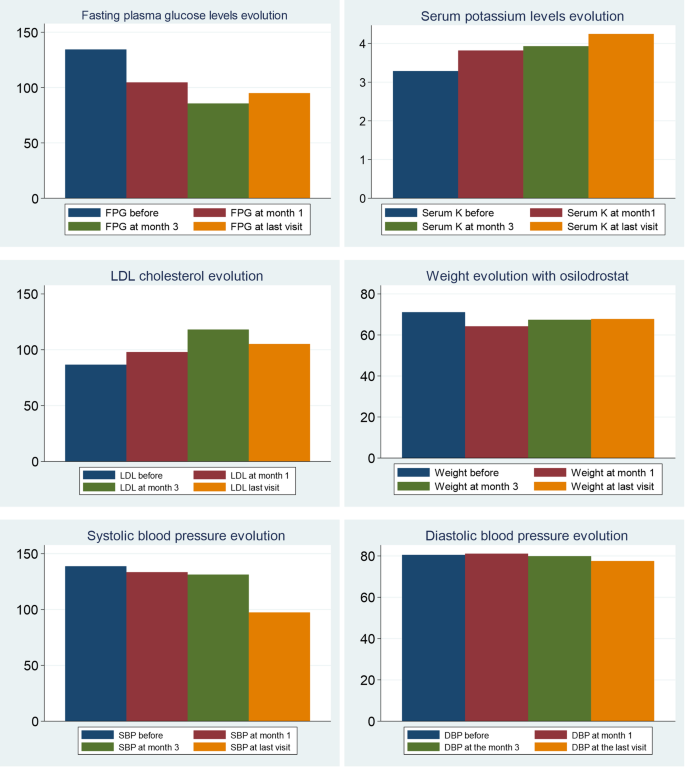
Examples of medications your doctor may prescribe for Cushing’s syndrome are: aminoglutethimide (eg, Cytadren), ketoconazole (eg, Nizoral), metyrapone (eg, Metopirone), and mitotane (eg, Lysodren). Your doctor will let you know what medication—or combination of medications—is right for you.
You may also need to take medication after surgery to remove a pituitary tumor or adrenal tumor. Your doctor will most likely prescribe a cortisol replacement medication. This medication helps provide the proper amount of cortisol in your body. An example of this type of medication is hydrocortisone (a synthetic form of cortisol).
Experiencing the full effects of the medication can take up to a year or longer. But in most cases and under your doctor’s careful supervision, you can slowly reduce your use of cortisol replacement medications because your body will be able to produce normal cortisol levels again on its own. However, in some cases, people who have surgery to remove a tumor that causes Cushing’s syndrome won’t regain normal adrenal function, and they’ll typically need lifelong replacement therapy.2
Treating Cushing’s Syndrome Conclusion
You may need one treatment or a combination of these treatments to effectively treat your Cushing’s syndrome. Your doctor will let you know what treatments for Cushing’s syndrome you’ll need.
From https://www.endocrineweb.com/conditions/cushings-syndrome/cushings-syndrome-treatments
Filed under: adrenal, Cushing's, pituitary, Treatments | Tagged: ACTH, adrenal, Adrenocorticotropic hormone, corticosteroids, cortisol, Cushing's Syndrome, ectopic, ketoconazole, Metyrapone, mitotane, pituitary, radiation, recurrence, surgery | Leave a comment »







-thorax-")

")

