Integration of Clinical Studies With Case Presentations
Maria Fleseriu, Richard J Auchus, Irina Bancos, Beverly MK Biller Journal of the Endocrine Society, Volume 9, Issue 4, April 2025, bvaf027 https://doi.org/10.1210/jendso/bvaf027
Abstract
Although most cases of endogenous Cushing syndrome are caused by a pituitary adenoma (Cushing disease), approximately one-third of patients present with ectopic or adrenal causes.
Surgery is the first-line treatment for most patients with Cushing syndrome; however, medical therapy is an important management option for those who are not eligible for, refuse, or do not respond to surgery.
Clinical experience demonstrating that osilodrostat, an oral 11β-hydroxylase inhibitor, is effective and well tolerated comes predominantly from phase III trials in patients with Cushing disease. Nonetheless, reports of its use in patients with ectopic or adrenal Cushing syndrome are increasing. These data highlight the importance of selecting the most appropriate starting dose and titration frequency while monitoring for adverse events, including those related to hypocortisolism and prolongation of the QT interval, to optimize treatment outcomes. Here we use illustrative case studies to discuss practical considerations for the management of patients with ectopic or adrenal Cushing syndrome and review published data on the use of osilodrostat in these patients.
The case studies show that to achieve the goal of reducing cortisol levels in all etiologies of Cushing syndrome, management should be individualized according to each patient’s disease severity, comorbidities, performance status, and response to treatment. This approach to osilodrostat treatment maximizes the benefits of effective cortisol control, leads to improvements in comorbid conditions, and may ameliorate quality of life for patients across all types and severities of Cushing syndrome.
This case report describes a 42-year-old female with a rare pheochromocytoma presenting without classic Cushingoid features but with uncontrolled hypertension, type 2 diabetes, and recurrent headaches. Despite the absence of typical signs, biochemical analysis revealed elevated cortisol and ACTH levels, and imaging showed a 6 cm adrenal mass. The patient was stabilized preoperatively with alpha-blockers and metyrapone before undergoing a successful laparoscopic adrenalectomy. Histopathology confirmed pheochromocytoma with aggressive features. Postoperatively, her blood pressure and symptoms improved, and her cortisol levels normalized. This case underscores the diagnostic challenges of ACTH-secreting pheochromocytomas without classic hypercortisolism signs and emphasizes the need for thorough endocrine and imaging assessments. Surgical resection remains the definitive treatment, with long-term follow-up essential to monitor for recurrence. This case contributes to the limited literature on the coexistence of pheochromocytoma and ectopic ACTH secretion.
Introduction
Ectopic ACTH-dependent tumors are rare, comprising approximately 5%–10% of Cushing syndrome cases, and are infrequently associated with pheochromocytomas, making this a unique presentation [1, 2]. Pheochromocytomas, though rare, can present as adrenal incidentalomas, often discovered during imaging for unrelated conditions. They represent 7% of adrenal incidentalomas and pose clinical challenges due to the risk of hormonal hypersecretion, including excess catecholamines and cortisol [1]. This case highlights the coexistence of an ectopic ACTH-producing tumor and pheochromocytoma, a combination rarely reported in the literature [3, 4]. While Cushing syndrome typically arises from adrenal or pituitary sources, ectopic ACTH secretion from pheochromocytomas presents a diagnostic and therapeutic challenge due to its rarity and aggressive potential [4–6]. Early diagnosis is crucial, particularly in cases with comorbidities like hypertension and diabetes, which are common in pheochromocytomas [1, 2]. This case underscores the need for a multidisciplinary approach to managing rare endocrine tumors.
Case report
A 42-year-old female from Mexico City presented with a history of treatment-resistant hypertension and a newly identified adrenal mass. She had no history of alcohol or tobacco use and led a generally healthy lifestyle. She was diagnosed with type 2 diabetes five years before symptoms appeared and developed hypertension five years before hospitalization, managed with valsartan and amlodipine verapamil.
The patient’s hypertension worsened, with blood pressure readings reaching 200/160 mmHg. She presented with asthenia and adynamia, and a CT scan revealed a 4 cm right adrenal mass, confirmed as 4.7 cm on a subsequent scan (Fig. 1). No signs of metastasis were observed. Upon hospital admission, her physical examination revealed a blood pressure of 95/84 mmHg, a heart rate of 95 beats per minute, a respiratory rate of 28 breaths per minute, and a systolic murmur. She exhibited no Cushingoid features.
Figure 1
The imaging identified a hyperdense area at the lower pole of the left kidney. A heterogeneous image was visualized in the right adrenal gland, characterized by a hypodense lesion measuring 40 × 47 × 43 mm, with a density of 36 Hounsfield units (HU) in the simple phase, 107 HU in the venous phase and 61 HU in the delayed phase (15 min), with an absolute washout of 64%.
Initial laboratory tests showed elevated white blood cells (11 000/mm3), hemoglobin of 12.5 g/dl, and platelet count of 305 000/mm3. Blood chemistry indicated hyperglycemia (132 mg/dl), hyponatremia (129 mEq/l), and hypokalemia (3.4 mEq/l). Cortisol levels were elevated at 31.53 μg/dl, and a 1 mg low-dose dexamethasone suppression test showed cortisol levels of 16.65 μg/dl and 14.63 μg/dl, suggesting ACTH-dependent Cushing syndrome.
ACTH levels were 24 pg/ml, which, while elevated, were not suppressed. However, elevated 24-h urinary metanephrines (9881 μg/24 h) confirmed the presence of pheochromocytoma. The patient’s aldosterone-to-renin ratio was measured, revealing a ratio of 4. The serum aldosterone level was 5 ng/dl (138 pmol/l), while plasma renin activity was recorded at 1.1 ng/ml/h.
Imaging revealed a 4.7 cm right adrenal mass with a density of 36 Hounsfield Units and an absolute washout of 64%, with no signs of malignancy (Fig. 1).
The patient’s hypertension was initially managed with prazosin and metoprolol, but her blood pressure spiked to 200/160 mmHg during a hypertensive crisis, requiring nitroprusside. Surgical intervention was planned after diagnosis was confirmed.
The patient underwent a successful laparoscopic right adrenalectomy. The tumor measured 6 cm, and histopathology confirmed a pheochromocytoma with a PASS score of 4, indicating potential for aggressive behavior (Table 1). Histological and immunohistochemical analysis revealed the tumor’s characteristic organoid pattern (Zellballen) with chromogranin and synaptophysin positivity in principal cells and S100 protein staining in sustentacular cells, consistent with pheochromocytoma (Fig. 2). Postoperatively, her blood pressure stabilized, and symptoms of palpitations and sweating resolved. She has weaned off antihypertensives, and a follow-up dexamethasone suppression test showed a significant reduction in cortisol levels (1.2 μg/dl), indicating successful tumor removal.
Specimen from right adrenalectomy:
Pheochromocytoma measuring 6×6 cm (positive for chromogranin 7, synaptophysin +S100, with sustentacular cells staining positive)
Marked nuclear pleomorphism: 1 point
Diffuse growth pattern: 2 points
Capsular invasion: 1 point
Total: 4 points.
Tumors with a score greater than 4 may exhibit aggressive biological behavior.
Figure 2
Histological and microscopic findings of adrenal Pheochromocytoma. (A) Macroscopic appearance. The ovoid tissue specimen has a light, smooth, soft external surface. The cut surface reveals a dark inner surface with light and hemorrhagic areas. Two cystic lesions with smooth walls are observed in the center (gross view). (B) A well-demarcated hypercellular lesion with an organoid pattern (Zellballen), separated by thin fibrovascular septa (Hematoxylin and eosin stain, 40×). (C) Nest of polygonal principal cells with ample eosinophilic granular cytoplasm, well-defined plasma membranes, hyperchromatic nuclei, and mild nuclear pleomorphism. Adjacent to the principal cells are spindle-shaped sustentacular cells with eosinophilic cytoplasm (Hematoxylin and eosin stain, 400×). (D) Positive immunoreactivity for chromogranin in principal cells. (E) Intense cytoplasmic reaction for synaptophysin in principal cells (immunohistochemistry, 400×). (F) Positive immunoreactivity for S100 protein, showing nuclear and cytoplasmic staining in sustentacular cells (immunohistochemistry, 400×).
Postoperatively, her course was uneventful, with stable blood pressure without antihypertensives. A follow-up evaluation revealed normal cortisol levels, and 24-h urinary metanephrines returned to normal (312 μg/24 h for metanephrines; 225 μg/24 h for normetanephrines). Repeat imaging showed no residual adrenal mass. At her most recent follow-up, the patient remained asymptomatic with normal laboratory values, and no recurrence has been detected.
Discussion
Ectopic ACTH-secreting pheochromocytomas are rare, accounting for a small percentage of ACTH-dependent Cushing syndrome cases [1, 4–6]. These tumors present diagnostic challenges, mainly when typical signs of Cushing syndrome, such as moon face, abdominal striae, or muscle weakness, are absent [3]. In this case, the patient exhibited only diabetes, uncontrolled hypertension, and recurrent headaches, symptoms that can also be attributed to pheochromocytoma itself [1]. The absence of Cushingoid features delayed the identification of ectopic ACTH secretion, making this case particularly difficult and unusual.
According to Gabi JN et al., most patients with ACTH-secreting pheochromocytomas present with severe hypercortisolism, including rapid weight gain and characteristic facial changes [3]. The absence of such features in this patient highlights the need to consider ectopic ACTH secretion in cases of adrenal masses, even without typical Cushing syndrome symptoms. This case illustrates how subtle presentations can lead to delayed diagnoses, emphasizing the importance of thorough evaluation in patients with adrenal tumors and metabolic abnormalities [1, 3].
The diagnostic approach for pheochromocytomas includes hormonal assays and imaging [7, 8]. Preoperative management for pheochromocytomas typically includes alpha-blockers to manage catecholamine excess [4, 7, 8]. This patient was managed with prazosin for blood pressure control and metyrapone to suppress cortisol production, consistent with clinical guidelines for managing ACTH-secreting tumors [5, 7, 8]. Despite the absence of Cushingoid features, careful preoperative preparation was essential to prevent complications during surgery.
Surgical resection is the definitive treatment for pheochromocytomas, particularly those secreting ACTH [8]. In this case, the patient underwent a successful laparoscopic adrenalectomy with no intraoperative complications. Histopathology confirmed a pheochromocytoma with marked nuclear pleomorphism and capsular invasion, suggesting potential aggressive behavior. Postoperatively, the patient’s blood pressure normalized, and her diabetes improved, aligning with outcomes reported in similar cases [4, 6]. Cortisol levels also returned to normal, demonstrating the effectiveness of adrenalectomy in resolving hypercortisolism.
A limitation in this case was the delayed recognition of ectopic ACTH secretion due to the absence of typical Cushingoid signs. The literature underscores the importance of considering this diagnosis, even in nonspecific cases [5].
Long-term management of pheochromocytomas, particularly those with aggressive features like capsular invasion, requires close follow-up [5, 7, 8]. Genetic testing should be considered, especially in patients with unusual presentations or family histories of endocrine disorders [1, 5]. Although not performed in this case, genetic testing could have provided further insight into the tumor’s etiology.
Acknowledgements
We thank the radiology department for interpreting the CT.
Conflict of interest
The authors declare no conflicts of interest related to this case report.
Funding
No external funding was received for this study.
Ethical approval
No approval was required.
Consent
Written informed consent was obtained from the patient and her parents to publish this case report and any accompanying images.
Guarantor
Froylan D. Martinez-Sanchez is the guarantor for this publication and accepts full responsibility for the work.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse, distribution, and reproduction in any medium, provided the original work is properly cited.
Neuroendocrine tumors (NETs) are heterogeneous neoplasms that arise from neuroendocrine cells, resulting in endocrine imbalances that impact quality of life and prognosis. Ectopic adrenocorticotropic hormone (ACTH) production by NETs is a rare cause of ACTH-dependent Cushing’s syndrome. While the majority of these cases are associated with intrathoracic tumors, recent reports have indicated an increasing incidence of cases originating from diverse anatomical sites. Furthermore, despite comprehensive imaging efforts, a substantial proportion of cases remain challenging to localize.
In this case, we describe a 54-year-old man with a stage IV NET with metastatic liver and pancreatic lesions, who presented with Cushing’s syndrome due to ectopic ACTH production. The patient exhibited symptoms of severe hypercortisolism, including weight gain, proximal muscle weakness, acute-onset heart failure, and hypertension. Imaging revealed bilateral adrenal hypertrophy. Laboratory tests revealed hypokalemia and hyperglycemia and confirmed elevated cortisol levels and a lack of suppression after dexamethasone administration, consistent with ectopic rather than pituitary ACTH production. The patient was treated with metyrapone because ketoconazole was contraindicated because of liver metastasis and recent upper gastrointestinal bleeding requiring proton pump inhibitor use. This case highlights the rare occurrence of ACTH-producing NETs and emphasizes the importance of considering this diagnosis in cases with similar presentations. Furthermore, medical management of this patient without surgical intervention, owing to multiple contraindications, offers an important perspective for treating complex cases.
Introduction
Neuroendocrine tumors (NETs) are a heterogeneous group of neoplasms that can secrete various hormones; however, ectopic adrenocorticotropic hormone (ACTH) production is rare, occurring in only 5-10% of all Cushing’s syndrome cases [1]. Liddle et al. described the first case in 1962 [2]. A recent case series that examined the clinical and diagnostic treatment of ectopic ACTH in a tertiary center included information on only 12 cases collected over a 17-year period [3]. The most common site for ectopic ACTH from malignancy is the intrathoracic region, primarily in small-cell lung carcinomas. Unfortunately, obtaining a single diagnostic image that can detect tumor-producing ACTH remains challenging. According to the literature, ectopic ACTH resulting in Cushing’s syndrome can remain undetected [3,4].
In the present case, a patient with a stage IV NET presented with the classic features of Cushing’s syndrome, leading to the diagnosis of ectopic ACTH production. The complexity of this case, owing to the patient’s metastatic disease, the contraindications for certain therapies, and the requirement for atypical medical management, highlights the challenges of treating advanced NETs, especially metastatic lesions with hormonal overproduction. This report aimed to underscore the importance of early recognition and the effectiveness of metyrapone as a treatment for hypercortisolism in metastatic NET.
Case Presentation
A 54-year-old man with a known history of a World Health Organization (WHO) grade 3, stage IV NET with metastatic lesions in the liver and pancreas presented to the hospital with new-onset acute heart failure. His medical history consisted of papillary thyroid cancer diagnosed in January 2023, for which he underwent total thyroidectomy and left neck dissection. Three months later, the patient was found to have a new liver lesion that was biopsied and was consistent with a WHO grade 3 NET (Figure 1). He was started on capecitabine and temozolomide chemotherapy regimen, which was switched to folinic acid, fluorouracil, and oxaliplatin due to disease progression. He had undergone positron emission tomography (PET)/computed tomography (CT) as part of the follow-up for NET, and the findings were consistent with hypermetabolic pancreatic and liver lesions. However, no uptake was observed in the lungs and/or adrenal glands (Figure 2).
Figure 1: Liver tissue section showing positive synaptophysin immunohistochemical staining in neoplastic cells, consistent with a neuroendocrine neoplasm.
Figure 2: FDG PET/CT scan of the whole body showing hypermetabolic pancreatic tail mass which measures up to 6.5 cm and multifocal liver hypermetabolic metastases.
The patient was admitted first with gastrointestinal (GI) bleeding secondary to duodenal ulcers that were managed with a proton pump inhibitor (PPI), pantoprazole 40 mg, oral, BID (Figure 3). Ten days later, he presented with worsening dyspnea and shortness of breath, and clinical examination was consistent with volume overload and 4+ pitting edema in the lower extremities. Additionally, he was found to have a significantly low potassium level (2.6 mmol/L) and worsening serum blood glucose (341 mg/dL). The constellation of symptoms in the patient, including significant weight gain, obesity, easy bruising, proximal muscle weakness, acute-onset heart failure, hypertension, hypokalemia, and worsening hyperglycemia with new insulin requirements, raised concerns about hypercortisolism and prompted testing. The serum ACTH levels were markedly elevated (488 pg/mL; reference range: 10-60 pg/mL). CT of the abdomen and pelvis revealed bilateral adrenal gland hypertrophy (Figure 4).
Figure 3: Upper endoscopy images showing four cratered, non-bleeding duodenal ulcers with a clean ulcer base (Forrest Class III).
Figure 4: CT of the abdomen and pelvis demonstrating bilateral adrenal gland hypertrophy.
CT: computed tomography
Morning cortisol levels were significantly increased (42.2 µg/dL), and the 8-mg dexamethasone suppression test showed no suppression, with a post-dexamethasone cortisol level of 44.2 µg/dL. The 24-hour urinary-free cortisol level was elevated (2259 µg/24 hour; reference range: 3.5-45 µg/24 hour). At this time, the differential diagnoses included but were not limited to Cushing’s disease or ectopic ACTH production secondary to metastatic NET. However, given that the patient had bilateral adrenal gland hypertrophy that was noted on imaging and his cortisol did not suppress with a high-dose dexamethasone suppression test, these findings support ectopic ACTH secretion secondary to metastatic NET over Cushing’s disease from a pituitary source.
After confirming the diagnosis, the patient was started on metyrapone 500 mg, administered two times per day; his serum cortisol began to decrease (from 42 to 38 µg/dL) and continued to decline until it reached the lowest level (8.9 µg/dL) with metyrapone 500 mg, administered four times per day. Unfortunately, because of cost-related issues, the patient was switched to octreotide; however, subsequently, his serum cortisol level increased (from 8.9 to 49 µg/dL). Ketoconazole was not a viable option because of drug-drug interactions with PPI. Alternative suppressive medications were considered and included osilodrostat and mifepristone. However, given the patient’s QTc prolongation and previous history of arrhythmia, it was felt that the use of these medications was too high risk for fatal arrhythmia. Given the limited medical options, the patient was evaluated for surgery, and, given the multiple comorbidities as well as metastatic disease without an apparent culprit lesion, he was not initially deemed to be a suitable surgical candidate. Therefore, metyrapone was reinitiated to control hypercortisolemia while the patient was admitted, and it effectively lowered his total serum cortisol levels. However, given that metyrapone was not a long-term option and medical management had failed (octreotide was ineffective in controlling serum cortisol levels, and ketoconazole could not be used due to drug-to-drug interactions with PPI), surgery was considered as an option. Despite the high risk associated with the procedures owing to the patient’s condition, bilateral adrenalectomy was performed, considering the lack of medical options and the patient’s goals of care. The patient was discharged home on oral hydrocortisone, 15 mg in the morning and 10 mg in the evening, in addition to fludrocortisone 0.1 mg daily. The patient’s body surface area is 2.5 m². The pathology of his adrenal glands was consistent with that of a metastatic NET (Figure 5). The patient was seen in the endocrinology clinic after bilateral adrenalectomy for a follow-up almost one month after the procedure. He reported feeling tired and falling asleep quite often. He used to be able to walk; however, now, he could only make it a quarter of the way due to muscle weakness. Unfortunately, further follow-up and outcome could not be evaluated as the patient died three months after his bilateral adrenalectomy surgery, and the cause of death was unknown.
Figure 5: Adrenal tissue section showing positive synaptophysin immunohistochemical staining in neoplastic cells, consistent with a neuroendocrine neoplasm.
Discussion
This case of a stage IV NET with ectopic ACTH production leading to Cushing’s syndrome is notable because of its rarity and complexity. Although NETs are known for their diverse hormonal secretions, only a small subset of them are associated with ACTH production, making this case an important addition to the limited literature.
NETs causing ectopic Cushing’s syndrome are most frequently found in the intrathoracic region (40-60%), including bronchial tumors, small-cell lung carcinoma, and thymic carcinomas. Additional sites where these tumors may occur include the pancreas and thyroid gland (particularly medullary thyroid carcinoma). Less common locations include the prostate, rectum, ovaries, and bladder [5].
Our patient’s PET/CT findings were consistent with those of hypermetabolic lesions in the liver and pancreas. However, there was no uptake in the lungs, which is the most common site reported in the literature [5]. Additionally, there was no uptake in the adrenal glands, and the pathology was later found to be consistent with NETs. This posed a challenge to the diagnosis and identification of the culprit lesion. Reportedly, high-resolution cross-sectional CT imaging has a sensitivity of 50-67% in identifying the source of ectopic ACTH production, and when the findings are negative, a variety of nuclear medicine functional imaging techniques (Octreoscan, fluorine-18 fluorodeoxyglucose PET/CT, and gallium-68 somatostatin receptor-targeted PET/CT) can be used [6]. However, despite advances in imaging modalities, up to 20% of ectopic ACTH syndrome cases remain occult after initial imaging [4,7].
ACTH-producing pancreatic neuroendocrine (pNE) tumors are rare malignancies characterized by their aggressive nature [8]. Individuals diagnosed with this condition have less favorable outcomes compared with those with insulinoma, gastrinoma, or nonfunctional ACTH-producing pNE tumors [9]. The underlying reasons for the aggressiveness of the tumor and the resulting poor patient outcomes remain elusive. One study proposed that decreased methylation of the proopiomelanocortin promoter may enhance the ability of the tumors to secrete ACTH [10].
A similar presentation was reported by Al-Toubah et al. in a 2023 case series on ACTH-secreting pNE neoplasms. That study highlighted the rarity of ACTH production in these tumors and emphasized that such cases often present with severe hypercortisolemia and Cushing’s syndrome. However, most patients in their series were treated with ketoconazole, which was not an option for our patient because of liver metastasis and recent upper GI bleeding requiring PPI treatment [11].
A systematic review published in February 2021 by Wu et al. investigated ACTH-producing pNE tumors. That study analyzed 210 publications, including data from 336 patients diagnosed with this condition. The results indicated a higher prevalence among female individuals (66.4%), at an average age of 44.7 years. The review reported the following frequencies of clinical symptoms: 69.3% experienced hypokalemia, 63.2% developed diabetes, 60.1% suffered from weakness, 56.4% had hypertension, 41.1% displayed moon face, and 37.4% presented with edema [12].
In the present case, the patient presented with decompensated heart failure, which is consistent with various case reports describing acute decompensated heart failure as the first presentation. Sugihara et al. reported three cases of Cushing’s syndrome characterized by left ventricular failure as the predominant feature associated with gross left ventricular hypertrophy [13]. Similarly, Petramala et al. reported a case of a 28-year-old woman with Cushing’s syndrome secondary to an adrenal adenoma who exhibited congestive heart failure as an initial symptom [14]. In this regard, some studies have examined the relationship between cardiac dysfunction and hypercortisolism and found that cardiac remodeling is independent of hypertension and is probably related to the direct action of cortisol on myocardial tissue via glucocorticoid receptors [15,16]. These cardiac impairments may be reversible with the appropriate treatment of the underlying hypercortisolism, such as the surgical resection of the adrenal adenoma or pituitary adenoma, and the medical management of heart failure [14].
Our patient received metyrapone and could not be treated using ketoconazole because of liver metastasis and drug-drug interactions with PPI, as previously mentioned. In 2022, Landry et al. studied the management of ACTH-secreting NETs [17]. Their study, including 76 patients, found that most patients had metastatic disease at the time of ectopic Cushing’s syndrome diagnosis, similar to our case. Furthermore, they found that de novo hyperglycemia predicted worse survival outcomes. Therefore, controlling the hypercortisolic phase is crucial. Unfortunately, most patients present with metastatic disease, which makes surgical management, that is, removing the ACTH-producing tumor, not always an option. Additionally, they found that patients with medically resistant ectopic Cushing’s syndrome, subsequently controlled with bilateral adrenalectomy, had significantly better disease-specific survival following ectopic Cushing’s syndrome diagnosis than did patients who did not undergo bilateral adrenalectomy.
In our case, there were limited treatment options given the metastatic burden and limitations in using some of the medications to control hypercortisolism. In their article, Landry et al. stated “We have learned this over time as, unfortunately, most patients in our cohort who were diagnosed with resistant ectopic Cushing syndrome only used one type of suppression therapy by the end of the study” [17]. One medication, peptide receptor radionuclide therapy, was reported in multiple studies [5,18,19]. However, the Food and Drug Administration did not approve this therapy until 2018, and it has not been examined for ectopic Cushing’s disease, especially in the metastatic NET setting.
As surgical resection remains the recommended first-line treatment for the majority of patients with Cushing’s syndrome [20], medical therapy plays a critical role when surgery is not feasible; many studies reviewed the use of agents such as mifepristone [21], levoketoconazole [22], and pasireotide [23,24]. Additionally, a recent review study that focused on the clinical consideration for osilodrostat in the management of patients with ectopic ACTH found that quality of life improved during the use of long-term osilodrostat as a treatment for ectopic Cushing’s syndrome raised from a pNE tumor [25].
Conclusions
This case highlights the complexities involved in the diagnosis and management of ectopic ACTH-producing NETs. Due to the rarity of such presentations, clinicians must maintain a high index of suspicion for ectopic ACTH production in patients with unexplained hypercortisolism, particularly when signs of Cushing’s syndrome are present. Additionally, the management of preoperative hypercortisolism may be challenging, as in our patient. The treatment approach in this case was unconventional, given the patient’s ineligibility for surgery due to difficulties in localizing the exact lesion and the metastatic disease. Medical management with metyrapone was chosen. However, as it was cost-prohibitive, alternative therapy with octreotide was attempted, but it failed to achieve adequate control. Ketoconazole was not an option given the recent GI bleeding, and eventually, our patient underwent bilateral adrenalectomy. Therefore, future studies are required to develop predictive markers to determine which patients will benefit from bilateral adrenalectomy versus long-term pharmacotherapy. An extensive study on perioperative management in cases with ectopic ACTH would have proven to be useful in ensuring the survival of our patient.
Thymic neuroendocrine tumor as a cause of Cushing syndrome is extremely rare in children.
Case presentation
We report a case of a 10-year-old girl who presented with typical symptoms and signs of hypercortisolemia, including bone fractures, growth retardation, and kidney stones. The patient was managed with oral ketoconazole, during which she experienced adrenal insufficiency, possibly due to either cyclic adrenocorticotropic hormone (ACTH) secretion or concurrent COVID-19 infection. The patient underwent a diagnostic work-up which indicated the possibility of an ACTH-secreting pituitary neuroendocrine tumor. However, after a transsphenoidal surgery, the diagnosis was not confirmed on histopathological examination. Subsequent bilateral inferior petrosal sinus sampling showed strong indications of the presence of ectopic ACTH syndrome. Detailed rereading of functional imaging studies, including 18F-FDG PET/MRI and 68Ga DOTATOC PET/CT, ultimately identified a small lesion in the thymus. The patient underwent videothoracoscopic thymectomy that confirmed a neuroendocrine tumor with ACTH positivity on histopathological examination.
Conclusion
This case presents some unique challenges related to the diagnosis, management, and treatment of thymic neuroendocrine tumor in a child. We can conclude that ketoconazole treatment was effective in managing hypercortisolemia in our patient. Further, a combination of functional imaging studies can be a useful tool in locating the source of ectopic ACTH secretion. Lastly, in cases of discrepancy in the results of stimulation tests, bilateral inferior petrosal sinus sampling is highly recommended to differentiate between Cushing disease and ectopic ACTH syndrome.
In children above seven years of age, the majority of pediatric Cushing syndrome (CS) cases are caused by a pituitary neuroendocrine tumors (PitNET). However, a differential diagnosis of hypercortisolemia in children is often challenging concerning the interpretation of stimulation tests and the fact that up to 50% of PitNET may not be detected on magnetic resonance imaging (MRI) [1]. An ectopic adrenocorticotropic hormone (ACTH) syndrome (EAS) is extremely rare in children. Its diagnosis is often missed or confused with Cushing disease (CD) [2]. Most ACTH-secreting tumors originate from bronchial or thymic neuroendocrine tumors (NETs), or less commonly, from NETs in other locations. To diagnose EAS, specific functional imaging studies are often indicated to elucidate the source of ACTH production.
Pharmacotherapy may be used before surgery to control hypercortisolemia and its symptoms/signs, or in patients in whom the source of hypercortisolism has not been found (e.g., EAS), or surgery failed. Ketoconazole or metyrapone, as adrenal steroidogenesis blockers, were found to be very efficient, although they exhibit side effects [3].
Furthermore, cyclic secretion of ACTH followed by fluctuating plasma cortisol levels is extremely rare in children, including those with EAS [4, 5]. Therefore, in cyclic EAS, the use of steroid inhibitors or acute illness or trauma can be associated with adrenal insufficiency, which can be life-threatening. Here we describe the clinical features, laboratory and radiological investigations, results, management, and clinical outcome of a 10-year-old girl with a thymic NET presenting with ACTH secretion.
Case presentation
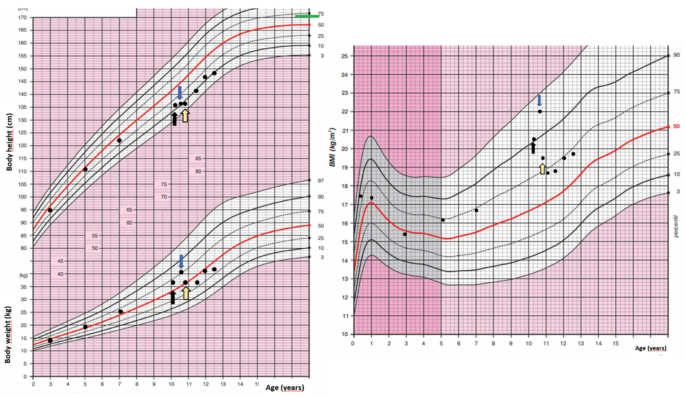
A 10-year-old girl was acutely admitted to our university hospital for evaluation of facial edema and macroscopic hematuria in May 2021. A day before admission, she presented to the emergency room for dysuria, pollakiuria, nausea, and pain in her right lower back. Over the past year she had experienced excessive weight gain with increased appetite and growth retardation (Fig. 1). Her height over three years had shifted from the 34th to the 13th centile (Fig. 1). Her parents noticed facial changes, pubic hair development, increased irritability, and moodiness.
Fig. 1
Body weight, body height, and body mass index development of the case patient. The black arrow indicates the first presentation, the blue arrow indicates the start of ketoconazole treatment and the yellow arrow indicates the time of thymectomy. Mid-parental height is indicated by the green line
At admission, she was found to have a moon face with a plethora, few acne spots on forehead, as well as facial puffiness. In contrast to slim extremities, an abnormal fat accumulation was observed in the abdomen. Purple striae were present on abdomen and thighs. She did not present with any bruising, proximal myopathy, or edema. On physical examination, she was prepubertal, height was 135 cm (13th centile), and weight was 37 kg (69th centile) with a BMI of 20.4 kg/m2 (90th centile). She developed persistent hypertension. Her past medical history was uneventful except for two fractures of her upper left extremity after minimal trips one and three years ago, both treated with a caste. Apart from hypothyroidism on the maternal side, there was no history of endocrine abnormalities or tumors in the family.
In the emergency room, the patient was started on sulfonamide, pain medication, and intravenous (IV) fluids. Her hypertensive crises were treated orally with angiotensin-converting enzyme inhibitor or with a combination of adrenergic antagonists and serotonin agonists administered IV. Hypokalemia had initially been treated with IV infusion and then with oral potassium supplements. A low serum phosphate concentration required IV management. The initial investigation carried out in the emergency room found hematuria with trace proteinuria. Kidney ultrasound showed a 5 mm stone in her right ureter with a 20 mm hydronephrosis. She did not pass any kidney stones, however, fine white sand urine analysis reported 100% brushite stone.
Hypercortisolemia was confirmed by repeatedly increased 24-hour urinary free cortisol (UFC), (5011.9 nmol/day, normal range 79.0-590.0 nmol/day). Her midnight cortisol levels were elevated (961 nmol/l, normal range 68.2–537 nmol/l). There was no suppression of serum cortisol after 1 mg overnight dexamethasone suppression test (DST) or after low-dose DST (LDDST). An increased morning plasma ACTH (30.9 pmol/l, normal range 1.6–13.9 pmol/) suggested ACTH-dependent hypercortisolemia. There was no evidence of a PitNET on a 1T contrast-enhanced MRI. The high-dose DST (HDDST) did not induce cortisol suppression (cortisol 1112 nmol/l at 23:00, cortisol 1338 nmol/l at 8:00). Apart from the kidney stone, a contrast-enhanced computed tomography (CT) of her neck, chest, and abdomen/pelvis did not detect any lesion. Various tumor markers were negative and the concentration of chromogranin A was also normal.
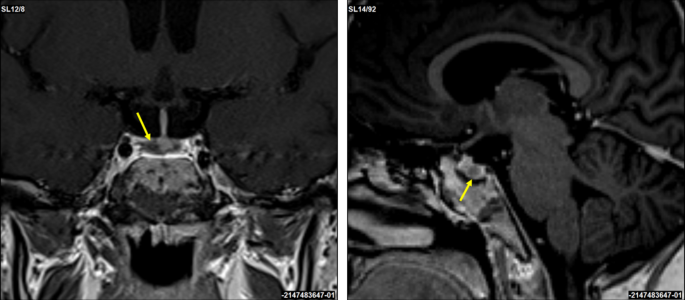
A corticotropin-releasing hormone (CRH) stimulation test induced an increase in serum cortisol by 32% at 30 min and ACTH concentration by 67% at 15 min (Table 1). A 3T contrast-enhanced MRI scan of the brain identified a 3 × 2 mm lesion in the lateral right side of the pituitary gland (Fig. 2). An investigation of other pituitary hormones was unremarkable. Apart from low serum potassium (minimal level of 2.8 mmol/l; normal range 3.3–4.7 mmol/l) and phosphate (0.94 mmol/l; normal range 1.28–1.82 mmol/l) concentrations, electrolytes were normal. The bone mineral density assessed by whole dual-energy X-ray absorptiometry was normal.
Fig. 2
Coronal and sagittal 3T contrast-enhanced brain MRI scans. A suspected 3 × 2 mm lesion in the lateral right side of the pituitary gland (yellow arrows)
The patient was presented at the multidisciplinary tumor board and it was decided that she undergoes transsphenoidal surgery for the pituitary lesion. No PitNET was detected on histopathological examination and no favorable biochemical changes were noted after surgery. After the patient recovered from surgery, subsequent bilateral inferior petrosal sinus sampling (BIPSS) confirmed EAS as the maximum ratio of central to peripheral ACTH concentrations was only 1.7. During the investigation for tumor localization, she was started on ketoconazole treatment (300 mg/day) to alleviate symptoms and signs of hypercortisolism. Treatment with ketoconazole had a beneficial effect on patient health (Fig. 1). There was a weight loss of 2 kg in a month, a disappearance of facial plethora, and a decrease in vigorous appetite. Her liver function tests remained within the normal range.
Table 1 Result of corticotropin-releasing hormone stimulation test
The 24-hour UFC excretion normalized three weeks after ketoconazole initiation. However, six weeks after continuing ketoconazole therapy (400 mg/day), the patient complained of nausea, vomiting, and diarrhea. She was found to have adrenal insufficiency with a low morning serum cortisol of 10.70 nmol/l (normal range 68.2–537 nmol/l) and salivary cortisol concentrations < 1.5 nmol/l (normal range 1.7–29 nmol/l). She was also found to be positive for COVID-19 infection. Ketoconazole treatment was stopped and our patient was educated to take stress steroids in case of persisting or worsening symptoms. Her clinical status gradually improved and steroids were not required.
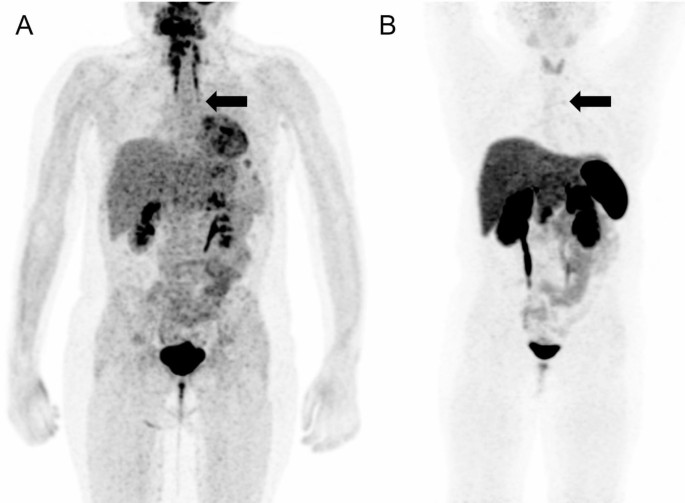
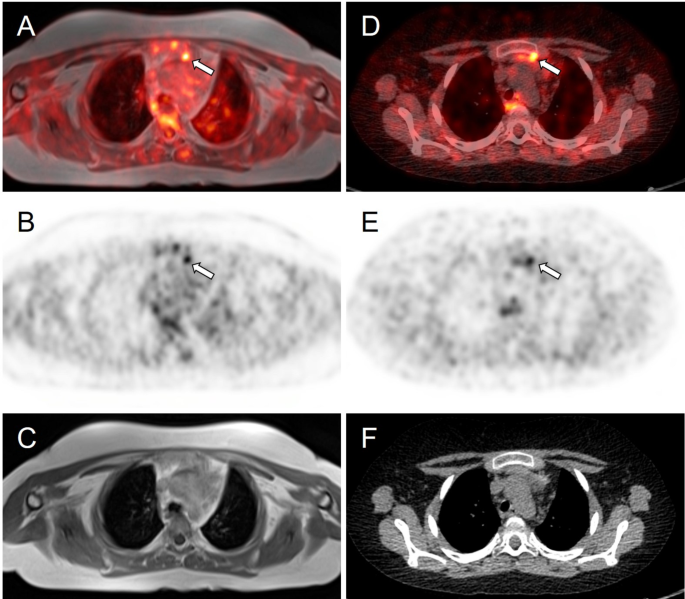
Meanwhile, whole-body fluorine-18 fluorodeoxyglucose positron emission tomography (18F-FDG PET)/MRI was performed with no obvious hypermetabolic lesion suspicious of a tumor. No obvious accumulation was detected on 68Ga-DOTATOC PET/CT images (Fig. 3). However, a subsequent careful and detailed re-review of the images detected a discrete lesion on 18F-FDG PET/MRI and 68Ga-DOTATOC PET/CT scans in the left anterior mediastinum, in the thymus (Fig. 4).
Fig. 3
18F-FDG PET/MRI (A) and 68Ga-DOTATOC (B) PET/CT scans. Whole body MIP reconstructions. Subtle correspondent focal hyperactivity in the left mediastinum (black arrow). The 18F-FDG PET/MRI image courtesy of Prof. Jiri Ferda, MD, PhD, Clinic of the Imaging Methods, University Hospital Plzen, Czech Republic
Axial slices of PET/MRI (A–C) and 68Ga-DOTATOC (D–F) PET/CT scans. Subtle correspondent focal hyperactivity in the left mediastinum (white arrow). No obvious finding on MRI (C) and CT (F) scans. The FDG PET/MRI image courtesy of Prof. Jiri Ferda, MD, PhD, Clinic of the Imaging Methods, University Hospital Plzen, Czech Republic
Three weeks after the episode of adrenal insufficiency and being off ketoconazole treatment, our patient´s pre-surgery laboratory tests showed slightly low morning cortisol 132 nmol/l with surprisingly normal ACTH 2.96 pmol/l (normal range 1.6–13.9 pmol/). Given the upcoming surgery, she was initiated on a maintenance dose of hydrocortisone (15 mg daily = 12.5 mg/m2/day). Further improvement of cushingoid characteristics (improvement of facial plethora and moon face, weight loss) was noticed. Our patient underwent videothoracoscopic surgery, and a hyperplastic thymus of 80 × 70 × 15 mm with a 4 mm nodule was successfully removed. Tumor immunohistochemistry was positive for ACTH, chromogranin A, CD56, and synaptophysin. Histopathological findings were consistent with a well-differentiated NET grade 1. A subsequent genetic screening did not detect any pathogenic variant in the MEN1 gene.
After surgery, hydrocortisone was switched to a stress dose and gradually decreased to a maintenance dose. Antihypertensive medication was stopped and further weight loss was observed after thymectomy. Within a few weeks after the thoracic surgery, the patient entered puberty, her mood improved significantly, and potassium supplements were stopped. Finally, hydrocortisone treatment was stopped ten months after thymectomy.
Discussion and conclusions
The case presented here demonstrates a particularly challenging work-up of the pediatric patient with the diagnosis of CS caused by EAS due to thymic NET. Differentiating CD and EAS can sometimes be difficult, including the use of various laboratory and stimulation tests and their interpretation, as well as proper, often challenging, reading of functional imaging modalities, especially if a discrete lesion is present at an unusual location [1]. When using established criteria for Cushing disease (for the CRH test an increase of cortisol and/or ACTH by ≥ 20% or ≥ 35%, respectively, and a ≥ 50% suppression of cortisol for the HDDST) our patient presented discordant results. The CRH stimulation test induced an increase in cortisol by 32% and ACTH by 67% and the 3T MRI pointed to the right-side pituitary lesion, both to yield false positive results. The HDDST, on the other hand, did not induce cortisol suppression and was against characteristic findings for CD. We did not proceed with desmopressin testing, which also induces an excess ACTH and cortisol response in CD patients and has rarely been used in pediatric patients, except in those with extremely difficult venous access [6]. Recently published articles investigated the reliability of CRH stimulation tests and HDDST and both concluded that the CRH test has greater specificity than HDDST [7, 8]. Elenius et al. suggested optimal response criteria as a ≥ 40% increase of ACTH and/or cortisol (cortisol as the most specific measure of CD) during the CRH test and a ≥ 69% suppression of serum cortisol during HDDST [7]. Using these criteria, the CD would be excluded in our patient. To demonstrate that the proposed thresholds for the test interpretation widely differ, Detomas et al. proposed a ≥ 12% cortisol increase and ≥ 31% ACTH increase during the CRH test to confirm CD [8].
The fact that up to 50% of PitNET may not be detected on MRI [1] and that more than 20% of patients with EAS are reported to have pituitary incidentalomas [9] makes MRI somewhat unreliable in differentiating CD and EAS. However, finally, well-established and generally reliable BIPSS in our patient supported the diagnosis of EAS. Thus, BIPSS is considered a gold standard to differentiate between CD and EAS; however, it can still provide false negative results in cyclic CS if performed in the trough phase [10] or in vascular anomalies or false positive results as in a recent case of orbital EAS [11].
In children, the presence of thymus tissue may be misinterpreted as normal. Among other reports of thymic NET [12], Hanson et al. reported a case of a prepubertal boy in whom a small thymic NET was initially treated as normal thymus tissue on CT [13]. In our case, initially, the lesion was not detected on the 18F-FDG and 68Ga-DOTATOC PET scans. A small thymic NET was visible only after a detailed and careful re-reading of both PET scans. Although somatostatin receptor (SSR) PET imaging may be helpful in identifying ectopic CRH- or ACTH-producing tumors, there are still some limitations [13]. For example, in the study by Wannachalee et al., 68Ga-DOTATATE identified suspected primary lesions causing ECS in 65% of patients with previously occult tumors and was therefore concluded as a sensitive method for primary as well as metastatic tumors [14]. In our patient, the final correct diagnosis was based on the results of both PET scans. This is in full support of the article published by Liu et al. who concluded that 18F-FDG and SSR PET scans are complementary in determining the proper localization of ectopic ACTH production [15]. Additionally, it is worth noting that not all NETs stain positively for ACTH which may present a burden in its identification.
To control hypercortisolemia, both ketoconazole and metyrapone were considered in our patient. Due to the side effects of metyrapone on blood pressure, ketoconazole was started as a preferred option in our pediatric patient. A retrospective multicenter study concluded that ketoconazole treatment is effective with acceptable side effects, with no fatal hepatitis and adrenal insufficiency in 5.4% of patients [3]. During ketoconazole treatment, our patient developed adrenal insufficiency; however, it is impossible to conclude whether this was solely due to ketoconazole treatment or whether an ongoing COVID-19 infection contributed to the adrenal insufficiency or whether this was caused by a phase of lower or no ACTH secretion from the tumor often seen in patients with cyclic ACTH secretion. The patient’s cyclic ACTH secretion is highly probable since her morning cortisol was slightly lower and ACTH was normal, even after being off ketoconazole treatment for 3 weeks.
When retrospectively and carefully reviewing all approaches to the diagnostic and management care of our pediatric patient, it would be essential to proceed to BIPSS before any pituitary surgery, especially when obtaining discrepant results from stimulation tests, as well as detecting a discrete pituitary lesion (≤ 6 mm) as recommended by the current guidelines [16]. This was our first experience using ketoconazole in a young child, and although this treatment was associated with very good outcomes in treating hypercortisolemia, close monitoring, and family education on signs and symptoms of adrenal insufficiency are essential to recognizing adrenal insufficiency promptly in any patient with EAS, especially those presenting also with some other comorbidities or stress, here COVID-19 infection.
In conclusion, the pediatric patient here presenting with EAS caused by thymic NET needs very careful assessment including whether cyclic CS is present, the outline of a good management plan to use all tests appropriately and in the correct sequence, monitoring carefully for any signs or symptoms of adrenal insufficiency, and apply appropriate imaging studies, with experienced radiologists providing accurate readings. Furthermore, ketoconazole treatment was found to be effective in reducing the symptoms and signs of CS in this pediatric patient. Finally, due to the rarity of this disease and the challenging work-up, we suggest that a multidisciplinary team of experienced physicians in CS management is highly recommended.
Data availability
No datasets were generated or analysed during the current study.
Streuli R. A rare case of an ACTH/CRH co-secreting midgut neuroendocrine tumor mimicking Cushing’s disease. Endocrinol Diabetes Metab Case Rep. 2017;2017:17–58. ,Krull I, Brändle M, et al.
Karageorgiadis AS, Papadakis GZ, Biro J, et al. Ectopic adrenocorticotropic hormone and corticotropin-releasing hormone co-secreting tumors in children and adolescents causing cushing syndrome: a diagnostic dilemma and how to solve it. J Clin Endocrinol Metab. 2015;100(1):141–8.
Mi Q, Yin M-Z, Gao Y-J et al. Thymic atypical carcinoid with cyclical Cushing’s syndrome in a 7-year-old boy: a case report and review of the literature. Intern Med. 2014;4(5).
Moszczyńska E, Pasternak-Pietrzak K, Prokop-Piotrkowska M, et al. Ectopic ACTH production by thymic and appendiceal neuroendocrine tumors – two case reports. J Pediatr Endocrinol Metab. 2020;34(1):141–6.
Crock PA, Ludecke DK, Knappe UJ, et al. A personal series of 100 children operated for Cushing’s disease (CD): optimizing minimally invasive diagnosis and transnasal surgery to achieve nearly 100% remission including reoperations. J Pediatr Endocrinol Metab. 2018;31(9):1023–31.
Detomas M, Ritzel K, Nasi-Kordhishti I, et al. Outcome of CRH stimulation test and overnight 8 mg dexamethasone suppression test in 469 patients with ACTH-dependent Cushing’s syndrome. Front Endocrinol (Lausanne). 2022;13:955945.
Yogi-Morren D, Habra MA, Faiman C, et al. Pituitary MRI findings in patients with pituitary and ectopic ACTH-dependent Cushing syndrome: does a 6-mm pituitary tumor size cut-off value exclude ectopic ACTH syndrome? Endocr Pract. 2015;21(10):1098–103.
Albani A, Berr CM, Beuschlein F, et al. A pitfall of bilateral inferior petrosal sinus sampling in cyclic Cushing’s syndrome. BMC Endocr Disord. 2019;19(1):105.
Tan H, Chen D, Yu Y, et al. Unusual ectopic ACTH syndrome in a patient with orbital neuroendocrine tumor, resulted false-positive outcome of BIPSS: a case report. BMC Endocr Disord. 2020;20(1):116.
Ahmed MF, Ahmed S, Abdussalam A, et al. A rare case of ectopic adrenocorticotropic hormone syndrome (EAS) in an adolescent girl with a thymic neuroendocrine tumour. Cureus. 2024;16(8):e66615.
Hanson JA, Sohaib SA, Newell-Price J, et al. Computed tomography appearance of the thymus and anterior mediastinum in active Cushing’s syndrome. J Clin Endocrinol Metab. 1999;84:602–5.
Wannachalee T, Turcu AF, Bancos I, et al. The clinical impact of [68 Ga]-DOTATATE PET/CT for the diagnosis and management of ectopic adrenocorticotropic hormone – secreting Tumours. Clin Endocrinol (Oxf). 2019;91(2):288–94.
Liu Q, Zang J, Yang Y, et al. Head-to-head comparison of 68Ga-DOTATATE PET/CT and 18F-FDG PET/CT in localizing tumors with ectopic adrenocorticotropic hormone secretion: a prospective study. Eur J Nucl Med Mol Imaging. 2021;48(13):4386–95.
Flesiriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing’s disease: a guideline update. Lancet Diabetes Endocrinol. 2021;9(12):847–75.
The authors thank all the colleagues from the Thomayer University Hospital and Military University Hospital who were involved in the inpatient care of this patient.
Funding
This work was supported by the Charles University research program Cooperatio Pediatrics, Charles University, Third Faculty of Medicine, Prague.
Author information
Authors and Affiliations
Department of Children and Adolescents, Third Faculty of Medicine, Charles University, University Hospital Kralovske Vinohrady, Šrobárova 50, Prague, 100 34, Czech Republic
Irena Aldhoon-Hainerová
Department of Pediatrics, Thomayer University Hospital, Prague, Czech Republic
Irena Aldhoon-Hainerová
Department of Medicine, Military University Hospital, Prague, Czech Republic
Mikuláš Kosák
Third Department of Medicine, First Faculty of Medicine, Charles University, Prague, Czech Republic
Michal Kršek
Institute of Nuclear Medicine, First Faculty of Medicine, Charles University, General University Hospital, Prague, Czech Republic
David Zogala
Developmental Endocrinology, Metabolism, Genetics and Endocrine Oncology Affinity Group, Eunice Kennedy Shriver NICHD, NIH, Bethesda, MD, USA
Karel Pacak
Contributions
All authors made individual contributions to the authorship. IAH, MK, MK, and DZ were involved in the diagnosis and management of this patient. DZ was responsible for the patient´s imaging studies. IAH wrote the first draft of the manuscript. KP revised the manuscript critically. All authors reviewed and approved the final draft.
Signed informed consent was obtained from the patient and the patient´s parents for the publication of this case report and accompanying images.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
Julie Lavoillotte, Kamel Mohammedi, Sylvie Salenave, Raluca Maria Furnica, Dominique Maiter, Philippe Chanson, Jacques Young, Antoine Tabarin The Journal of Clinical Endocrinology & Metabolism, Volume 109, Issue 11, November 2024, Pages 2882–2891 https://doi.org/10.1210/clinem/dgae258
Abstract
Context
Current guidelines for distinguishing Cushing’s disease (CD) from ectopic ACTH secretion (EAS) are questionable, as they use pituitary magnetic resonance imaging (MRI) as first-line investigation for all patients. CRH testing is no longer available, and they suggest performing inferior petrosal sinus sampling (BIPPS), an invasive and rarely available investigation, in many patients.
Objective
To establish noninvasive personalized diagnostic strategies based on the probability of EAS estimated from simple baseline parameters.
Design
Retrospective study.
Setting
University hospitals.
Patients
Two hundred forty-seven CD and 36 EAS patients evaluated between 2001 and 2023 in 2 French hospitals. A single-center cohort of 105 Belgian patients served as external validation.
Results
Twenty-four-hour urinary free cortisol (UFC) had the highest area under the receiver operating characteristic curve for discrimination of CD from EAS (.96 [95% confidence interval (CI), .92–.99] in the primary study and .99 [95% CI, .98–1.00] in the validation cohort). The addition of clinical, imaging, and biochemical parameters did not improve EAS prediction over UFC alone, with only BIPPS showing a modest improvement (C-statistic index .99 [95% CI, .97–1.00]). Three groups were defined based on baseline UFC: < 3 (group 1), 3–10 (group 2), and > 10 × the upper limit of normal (group 3), and they were associated with 0%, 6.1%, and 66.7% prevalence of EAS, respectively. Diagnostic approaches performed in our cohort support the use of pituitary MRI alone in group 1, MRI first followed by neck-to-pelvis computed tomography scan (npCT) when negative in group 2, and npCT first followed by pituitary MRI when negative in group 3. When not combined with the CRH test, the desmopressin test has limited diagnostic value.
Conclusion
UFC accurately predicts EAS and can serve to define personalized and noninvasive diagnostic algorithms.








.")



{kind=link}
{kind=link}