ABSTRACT
KEYWORDS
INTRODUCTION
CASE PRESENTATION

FIGURE 1. Timeline of notable events. Timeline summarizing key events including clinical course, treatments, and relapses.
TABLE 1. Clinical and biochemical features of ICS and SAI in the patient
| Empty Cell | Clinical Findings | Interpretation |
|---|---|---|
| Growth and development | Height: 143.5 cm (3rd percentile); mid-parental height: 171 ± 8 cm | Growth deceleration likely related to chronic glucocorticoid exposure and ICS |
| Weight and body composition | Weight: 53.3 kg (75th–90th percentile); BMI: 25.8 kg/m²; central obesity | Suggestive of glucocorticoid-induced lipogenesis and altered fat distribution |
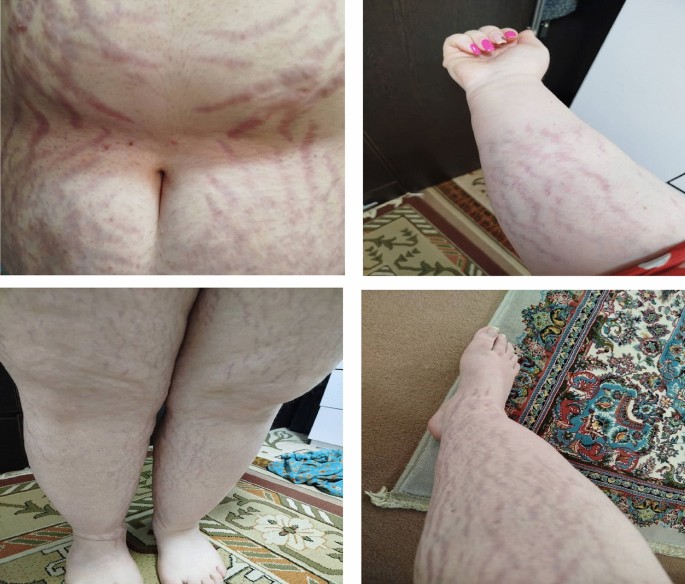
| Skin and soft tissue | Striae rubrae on flanks; mild dorsal fat pad (“buffalo hump”) | Classic phenotypic features of ICS |
| Pubertal status | Tanner stage I; testicular volume 5–6 mL; pubic hair stage I | Early puberty with preserved testicular volume; no signs of delayed or precocious puberty |
| HPA axis function | Cortisol: 0.5 → 9.9 → 3.1 µg/dL; ACTH: 7–23 pg/mL | Suppressed HPA axis consistent with SAI |
| Glucose metabolism | HbA1c: 5.9%; fasting glucose: 72 mg/dL; insulin: 16.9 mcU/mL | Normal glucose metabolism; mild hyperinsulinemia possibly due to steroid exposure |
| Thyroid function | TSH: 2.32 µU/mL; free T4: 1.59 ng/dL | Euthyroid; no evidence of central or primary thyroid dysfunction |
| Neurologic imaging | Right thalamic signal abnormality; stable; no neurological deficits | No CNS involvement of VKH; imaging excluded alternative diagnoses |
| Family history | Autoimmune conditions in maternal relatives; vitiligo in grandfather | Suggests genetic predisposition to autoimmune diseases; relevant to VKH etiology |
| Therapeutic course | Initial improvement with prednisone; relapses on tapering; mycophenolate added; steroids reintroduced | Demonstrates difficulty in achieving steroid-free remission and the need for steroid-sparing agents |
DISCUSSION

FIGURE 2. Multidisciplinary management plan for pediatric VKH with chronic corticosteroid therapy. Schematic representation of the recommended multidisciplinary team for pediatric patients with VKH requiring prolonged corticosteroid therapy. The model emphasizes collaboration among health professionals for early recognition and management of VKH manifestations.
CONCLUSION
REPORTING CHECKLIST DISCLOSURE
DATA AVAILABILITY STATEMENT
FUNDING
PATIENT CONSENT
ETHICAL STATEMENTS
CRediT authorship contribution statement
CONFLICTS OF INTEREST
REFERENCES
- Abu El-Asrar et al., 2008
A.M. Abu El-Asrar, A.S. Al-Kharashi, H. Aldibhi, H. Al-Fraykh, D. KangaveVogt-Koyanagi-Harada disease in childrenEye (London, England), 22 (9) (2008), pp. 1124-1131, 10.1038/sj.eye.6702859
- Abu El-Asrar et al., 2021
A.M. Abu El-Asrar, J. Van Damme, S. Struyf, G. OpdenakkerNew perspectives on the immunopathogenesis and treatment of uveitis associated with vogt-koyanagi-harada diseaseFrontiers in Medicine, 8 (2021), Article 705796, 10.3389/fmed.2021.705796
- Albaroudi et al., 2020
N. Albaroudi, M. Tijani, N. Boutimzine, O. CherkaouiClinical and therapeutic features of pediatric Vogt-Koyanagi-Harada diseaseJournal Français d’Ophtalmologie, 43 (5) (2020), pp. 427-432, 10.1016/j.jfo.2019.10.005
- Arroyo et al., 2023
R.G.G. Arroyo, H.I.P. Belmontes, M.D.J.G. Torres, B.M.R. Mendoza, B.E. Gonzalez, N.C. Bonilla, A.A.R. Ceja, G.G. SantiagoDrug induced iatrogenic Cushing’s syndromeInternational Journal of Research in Medical Sciences, 12 (1) (2023), pp. 303-308, 10.18203/2320-6012.ijrms20234026
- Beuschlein et al., 2024
F. Beuschlein, T. Else, I. Bancos, S. Hahner, O. Hamidi, L. Van Hulsteijn, E.S. Husebye, N. Karavitaki, A. Prete, A. Vaidya, C. Yedinak, O.M. DekkersEuropean society of endocrinology and endocrine society joint clinical guideline: diagnosis and therapy of glucocorticoid-induced adrenal insufficiencyEuropean Journal of Endocrinology, 190 (5) (2024), pp. G25-G51, 10.1093/ejendo/lvae029
- Bornstein et al., 2016
S.R. Bornstein, B. Allolio, W. Arlt, A. Barthel, A. Don-Wauchope, G.D. Hammer, E.S. Husebye, D.P. Merke, M.H. Murad, C.A. Stratakis, D.J. TorpyDiagnosis and treatment of primary adrenal insufficiency: an endocrine society clinical practice guidelineThe Journal of Clinical Endocrinology and Metabolism, 101 (2) (2016), pp. 364-389, 10.1210/jc.2015-1710
- Broersen et al., 2015
L.H.A. Broersen, A.M. Pereira, J.O.L. Jørgensen, O.M. DekkersAdrenal insufficiency in corticosteroids use: systematic review and meta-analysisThe Journal of Clinical Endocrinology and Metabolism, 100 (6) (2015), pp. 2171-2180, 10.1210/jc.2015-1218
- Herbort and Mochizuki, 2007
C.P. Herbort, M. MochizukiVogt-Koyanagi-Harada disease: Inquiry into the genesis of a disease name in the historical context of Switzerland and JapanInternational Ophthalmology, 27 (2–3) (2007), pp. 67-79, 10.1007/s10792-007-9083-4
- Improda et al., 2024
N. Improda, L. Chioma, D. Capalbo, C. Bizzarri, M. SalernoGlucocorticoid treatment and adrenal suppression in children: current view and open issuesJournal of Endocrinological Investigation, 48 (1) (2024), pp. 37-52, 10.1007/s40618-024-02461-9
- Leal et al., 2024
I. Leal, L.R. Steeples, S.W. Wong, C. Giuffrè, S. Pockar, V. Sharma, E.K.Y. Green, J. Payne, N.P. Jones, A.S.E. Chieng, J. AshworthUpdate on the systemic management of noninfectious uveitis in children and adolescentsSurvey of Ophthalmology, 69 (1) (2024), pp. 103-121, 10.1016/j.survophthal.2023.01.002
- Martin et al., 2010
T.D. Martin, S.R. Rathinam, E.T. CunninghamPrevalence, clinical characteristics, and causes of vision loss in children with Vogt-Koyanagi-Harada disease in South IndiaRetina (Philadelphia, Pa.), 30 (7) (2010), pp. 1113-1121, 10.1097/IAE.0b013e3181c96a87
- Messazos and Zacharin, 2016
B.P. Messazos, M.R. ZacharinLessons from iatrogenic Cushing syndrome in childrenJournal of Paediatrics and Child Health, 52 (12) (2016), pp. 1106-1110, 10.1111/jpc.13273
- Oo et al., 2020
Ei Ei Lin Oo, S.-P. Chee, K.K.Y. Wong, Hla Myint HtoonVogt-Koyanagi-Harada disease managed with immunomodulatory therapy within 3 months of disease onsetAmerican Journal of Ophthalmology, 220 (2020), pp. 37-44, 10.1016/j.ajo.2020.07.036
- Park et al., 2022
H.S. Park, H.Y. Park, C.S. Lee, S.C. Lee, J.H. LeeEfficacy of combined systemic corticosteroid and early immunomodulatory therapy within three months of onset in Vogt-Koyanagi-Harada diseaseRetina (Philadelphia, Pa.), 42 (12) (2022), pp. 2361-2367, 10.1097/IAE.0000000000003617
- Prete and Bancos, 2021
A. Prete, I. BancosGlucocorticoid induced adrenal insufficiencyBMJ (2021), p. n1380, 10.1136/bmj.n1380
- Reiff, 2020
A. ReiffClinical presentation, management, and long-term outcome of pars Planitis, Panuveitis, and Vogt-Koyanagi-Harada disease in children and adolescentsArthritis Care & Research, 72 (11) (2020), pp. 1589-1596, 10.1002/acr.24056
- Sadhu et al., 2024
S. Sadhu, P. Dutta Majumder, M. Shah, R. GeorgeVogt-Koyanagi-Harada Disease in Pre-school ChildrenOcular Immunology and Inflammation, 32 (4) (2024), pp. 415-418, 10.1080/09273948.2022.2117707
- Sakata et al., 2015
V.M. Sakata, F.T. Da Silva, C.E. Hirata, M.L.C. Marin, H. Rodrigues, J. Kalil, R.A. Costa, J.H. YamamotoHigh rate of clinical recurrence in patients with Vogt–Koyanagi–Harada disease treated with early high-dose corticosteroidsGraefe’s Archive for Clinical and Experimental Ophthalmology, 253 (5) (2015), pp. 785-790, 10.1007/s00417-014-2904-z
- Santos-Oliveira et al., 2025
J. Santos-Oliveira, L. Torrão, S. Torres-Costa, M. Silva, A.C. Pedrosa, J. Araújo, L. Figueira, C. Oliveira-FerreiraIatrogenic Cushing syndrome and secondary adrenal insufficiency in a child due to topical ocular corticosteroids: a case reportCase Reports in Ophthalmology, 16 (1) (2025), pp. 194-201, 10.1159/000543908
- Savage and Storr, 2012
M.O. Savage, H.L. StorrPediatric Cushing’s disease: Management IssuesIndian Journal of Endocrinology and Metabolism, 16 (Suppl 2) (2012), pp. S171-S175, 10.4103/2230-8210.104032
- Simonini et al., 2013
G. Simonini, P. Paudyal, G.T. Jones, R. Cimaz, G.J. MacfarlaneCurrent evidence of methotrexate efficacy in childhood chronic uveitis: A systematic review and meta-analysis approachRheumatology (Oxford, England), 52 (5) (2013), pp. 825-831, 10.1093/rheumatology/kes186
- Sood and Angeles-Han, 2017
A.B. Sood, S.T. Angeles-HanAn update on treatment of pediatric chronic non-infectious uveitisCurrent Treatment Options in Rheumatology, 3 (1) (2017), pp. 1-16, 10.1007/s40674-017-0057-z
- Stern and Nataneli, 2025
E.M. Stern, N. NataneliVogt-Koyanagi-Harada SyndromeStatPearls, StatPearls Publishing (2025)
- Wang et al., 2023
K. Wang, C. Zheng, G. Zhao, M. Zhang, T. Liu, H. Li, Q. Tao, Z. Cheng, X. Li, X. ZhangHigh long-term drug-free remission rate for acute vogt-koyanagi-harada disease with an appropriate immunosuppressive regimenRetina (Philadelphia, Pa.), 43 (9) (2023), pp. 1496-1505, 10.1097/IAE.0000000000003837
- Yang et al., 2023
P. Yang, W. Liao, Y. Pu, Z. Zhong, H. Wang, Q. Yu, J. Cai, W. Wang, G. SuVogt-Koyanagi-Harada disease in pediatric, adult and elderly: Clinical characteristics and visual outcomesGraefe’s Archive for Clinical and Experimental Ophthalmology, 261 (9) (2023), pp. 2641-2650, 10.1007/s00417-023-06058-5
Filed under: Cushing's, Rare Diseases, symptoms | Tagged: corticosteroids, growth delay, iatrogenic, Iatrogenic Cushing's Syndrome, mycophenolate mofetil, secondary adrenal insufficiency, striae, Vogt-Koyanagi-Harada disease (VKH), Weight gain | Leave a comment »