


After 8 years of menopause-like symptoms, a 62 year old patient discovered she actually had Cushing’s disease. Read about her long journey to get a diagnosis and her success in finding her dream-come-true neurosurgeon.
A Mysterious Decline in Health
When Elisabeth N., 62, started developing symptoms that included obesity, osteoporosis, insomnia, kidney stones and hair loss, she attributed it to what most women her age would: menopause. Back in 2000 she never would have thought those seemingly normal symptoms for a woman her age would lead her to Santa Monica to be treated eight years later by Daniel Kelly, MD., neurosurgeon and director of the Pacific Pituitary Disorders Center at Pacific Neuroscience Institute and Saint John’s Health Center. In fact, it wasn’t till February of 2008 that she learned it could all be caused by something completely different.
A Sister’s Observation Leads to a Breakthrough
“I wouldn’t have known about Cushing’s disease if it weren’t for my youngest sister; I’m 25 years older and so fortunate she has her medical degree,” explained Elisabeth, a kitchen and bath designer in Mesa, Arizona. “We hadn’t seen one another for five years when we visited in February of 2008. My appearance had drastically changed by then. She told me, ‘Don’t be scared, Bethie, but I think you should be tested for Cushing’s.’”
Learning About Cushing’s Disease
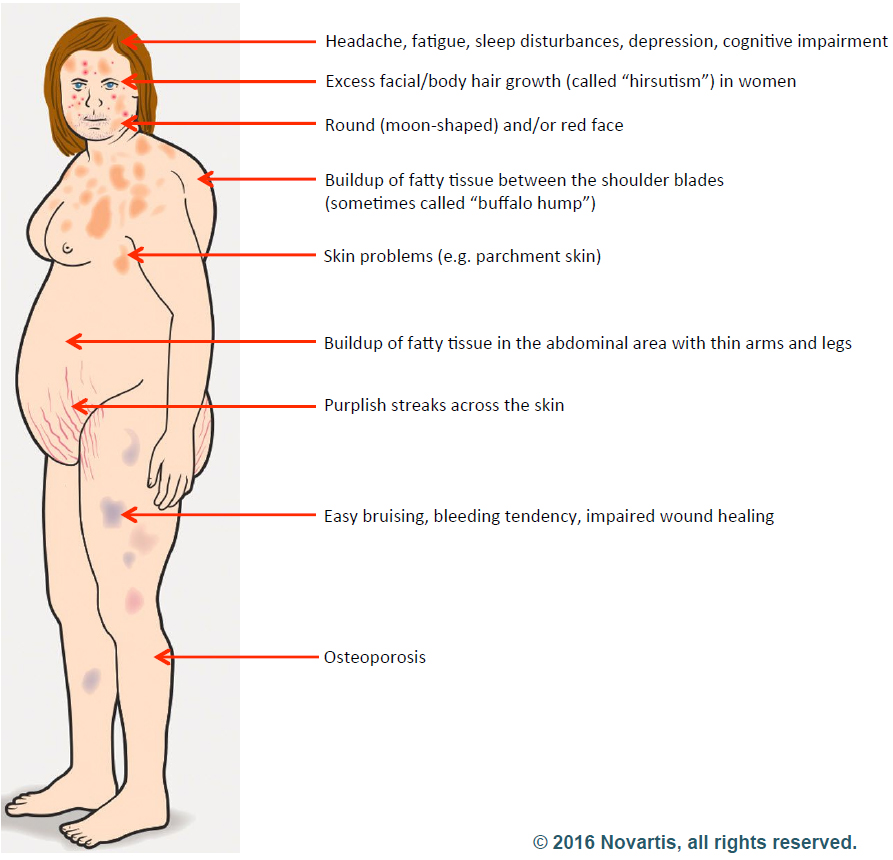
Elisabeth started researching Cushing’s disease right away and was relieved to learn that a cure was possible. Cushing’s is a hormonal disorder caused by high levels of the hormone cortisol. Symptoms include upper-body obesity, fragile skin that bruises easily, weakened bones, severe fatigue, weak muscles, high blood pressure, high blood glucose, increased thirst and urination, depression and a fatty hump between the shoulders. Women can also experience irregular menstrual periods and excess hair growth on their bodies. It can be caused by taking glucocorticoids such as prednisone or if there is a problem with a person’s pituitary gland or hypothalamus.
A Long and Uncertain Testing Journey
Elisabeth immediately set up a doctor’s appointment to get her cortisol and adrenocorticotropic hormone (ACTH – the pituitary hormone that stimulates the adrenal glands to make cortisol) levels tested. Over the next nine months Elisabeth went through several blood, urine, saliva and plasma tests for her cortisol and ACTH production and had an MRI. The tests showed elevated cortisol and ACTH levels but the initial impression was that her levels were not high enough to indicate Cushing’s disease and her pituitary MRI showed no apparent tumor. Elisabeth met with both a pituitary neurosurgeon and an endocrinologist, but both determined her condition not to be Cushing’s.
“My cortisol was not high enough; I wasn’t obese enough; I wasn’t disabled enough; I wasn’t depressed enough,” Elisabeth said.
Finding the Missing Clue
She felt frustrated by the diagnosis and continued to research possibilities online. It was during this research that she stumbled upon an article comparing MRI Tesla strengths. It recommended getting a Tesla 3.0 with contrast to pick up small abnormalities. Elisabeth scheduled a new MRI at the beginning of November. This time the scan detected a 6 mm tumor on the right side of her pituitary gland. Additional hormonal testing also confirmed that she did indeed have high ACTH and cortisol levels consistent with Cushing’s, “I was thrilled to finally have proof I had Cushing’s, but terrified because I knew I’d have to have brain surgery to remove it,” Elisabeth said. “I knew I wanted transsphenoidal surgery – the safest, most successful procedure with the least complications if done by an experienced surgeon.”
Understanding the Endonasal Transsphenoidal Approach
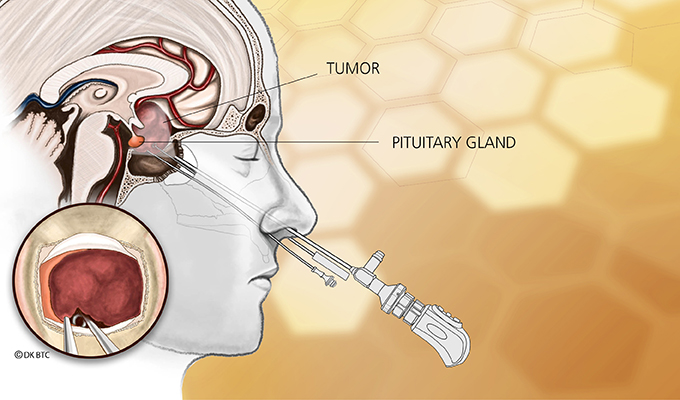
Endonasal transsphenoidal tumor removal, aka endoscopic endonasal approach, such as what Elisabeth needed, is a surgery that uses the nostril as the entry point with visualization from the operating microscope and endoscope. The approach passes through the back of the nasal cavity and into the sphenoid sinus to the skull base without facial incisions, brain retraction or post-operative nasal packing.
“This type of surgery is ideal for removing over 99% of pituitary adenomas, like what Elisabeth had and is considered first-line therapy for patients with Cushing’s disease,” Dr. Kelly explained. “Untreated or incompletely treated, Cushing’s disease is a very serious condition leading to uncontrolled hypertension, diabetes, weight gain and increased mortality.”
Choosing the Right Surgeon
With all the months she had to prepare for a diagnosis, Elisabeth knew exactly what needed to be done.
“I’d known I wanted Dr. Kelly to perform surgery but never imagined it could happen,” Elisabeth said. “I found him online. He’s ranked as one of the world’s top neurosurgeons specializing in this type of surgery. While watching his YouTube videos, I was awed by his kindness, patience, sense of humor, approachability, professionalism and complete lack of arrogance in spite of his fame. I’m still pinching myself that I had the fortune and honor to become one of his patients.”
A Life-Changing Call
Figuring she didn’t have anything to lose, Elisabeth called Dr. Kelly’s office and asked if he was accepting new patients (he was) and how long was his waiting list (she could see him next week). Elisabeth was astounded. She immediately mailed Dr. Kelly all the test results, films and reports she could gather.
“Two days later, Dr. Kelly personally called and left a message, indicating it appeared there was indeed an ACTH secreting adenoma on my pituitary gland and to call him back,” Elisabeth said. “I was blown away. I’d have expected to win the lottery first.”
Successful Surgery and a New Beginning
Dr. Kelly arranged to perform Elisabeth’s surgery two weeks later on November 26 – the day before Thanksgiving. Her cortisol levels fell dramatically within 24 hours of surgery. She has remained in remission since then. Years after surgery, she continues to feel like a new person and regularly stays in contact with Dr. Kelly and his office staff.
About Dr. Daniel Kelly

Dr. Daniel Kelly, a board-certified neurosurgeon, is the director and one of the founders of the Pacific Neuroscience Institute, director of the Pacific Brain Tumor Center and Pacific Pituitary Disorders Center, and is Professor of Neurosurgery at Saint John’s Cancer Institute at Providence Saint John’s Health Center. Considered to be one of the top neurosurgeons in the US, he is a multiple recipient of the Patients’ Choice Award and Southern California Super Doctors distinction.
Filed under: Cushing's, General Public, pituitary, symptoms, Treatments | Tagged: Cushing's, Dr. Daniel Kelly, endonasal, menopause, Pacific Brain Tumor Center, pituitary, Transsphenoidal surgery | Leave a comment »
