Abstract
Summary
This case report describes a rare presentation of ectopic Cushing’s syndrome (CS) due to ectopic corticotropin-releasing hormone (CRH) production from a medullary thyroid carcinoma (MTC).
The patient, a 69-year-old man, presented with symptoms of muscle weakness, facial plethora, and easy bruising.
An inferior petrosal sinus sampling test (IPSS) demonstrated pituitary adrenocorticotrophic hormone (ACTH) secretion, but a whole-body somatostatin receptor scintigraphy (68Ga-DOTATOC PET/CT) revealed enhanced uptake in the right thyroid lobe which, in addition to a grossly elevated serum calcitonin level, was indicative of an MTC. A 18F-DOPA PET/CT scan supported the diagnosis, and histology confirmed the presence of MTC with perinodal growth and regional lymph node metastasis.
On immunohistochemical analysis, the tumor cell stained positively for calcitonin and CRH but negatively for ACTH. Distinctly elevated plasma CRH levels were documented. The patient therefore underwent thyroidectomy and bilateral adrenalectomy.
This case shows that CS caused by ectopic CRH secretion may masquerade as CS due to a false positive IPSS test. It also highlights the importance of considering rare causes of CS when diagnostic test results are ambiguous.
Learning points
- Medullary thyroid carcinoma may secrete CRH and cause ectopic CS.
- Ectopic CRH secretion entails a rare pitfall of inferior petrosal sinus sampling yielding a false positive test.
- Plasma CRH measurements can be useful in selected cases.
Background
The common denominator of Cushing’s syndrome (CS) is autonomous hypersecretion of cortisol (1) and it is subdivided into ACTH-dependent and ACTH-independent causes. The majority of CS cases are ACTH-dependent (80–85%) with a pituitary corticotroph tumor as the most prevalent cause (Cushing’s disease), and less frequently an ectopic ACTH-producing tumor (2). The gold standard method to ascertain the source of ACTH secretion in CS patients is inferior petrosal sinus sampling (IPSS) with measurement of plasma ACTH levels in response to systemic corticotropin-releasing hormone (CRH) stimulation (3). The IPSS has a very high sensitivity and specificity of 88–100% and 67–100%, respectively (4), but pitfalls do exist, including the rare ectopic CRH-producing tumor, which may yield a false positive test result (3). Here, we describe a very rare case masquerading as CS including a positive IPSS test.
Case presentation
A 69-year-old man presented at a local hospital with a 6-month history of progressive fatigue, muscle weakness and wasting, easy bruising, facial plethora, and fluid retention. His serum potassium level was 2.6 mmol/L (reference range: 3.5–4.2 mmol/L) without a history of diuretics use. His previous medical history included spinal stenosis, benign prostatic hyperplasia, and hypertension. An electromyography showed no sign of polyneuropathy and an echocardiography showed no signs of heart failure with an ejection fraction of 55%. MRI of the spine revealed multiple compression fractures, and the patient underwent spinal fusion and decompression surgery; during this admission he was diagnosed with type 2 diabetes (HbA1c: 55 mmol/mol). After spine surgery, the patient developed a pulmonary embolism and initiated treatment with rivaroxaban.
Establishing the diagnosis of ACTH-dependent CS
Six months after his spine surgery, the patient was referred to the regional department of endocrinology for osteoporosis management. Blood tests revealed a low serum testosterone level with non-elevated luteinizing hormone (LH) and follicle-stimulating hormone (FSH) levels (Table 1). An overnight 1 mg dexamethasone suppression test was positive with a morning cortisol level of 254 nmol/L and three consecutive 24-h urinary cortisol levels were markedly elevated with mean level of ≈600 nmol/24 h (reference range: 12–150 nmol/24 h). A single plasma ACTH was 37 ng/L (Table 1).
| Parameters | Patient’s values | Reference range |
|---|---|---|
| ACTH, ng/L | 37 | 7–64 |
| UFC, nmol/day | 588 | 12–150 |
| Urinary cortisol, nmol/L | 600 | 171–536 |
| OD, nmol/L | 254 | <50 |
| Free testosterone, nmol/L | 0.061 | 0.17–0.59 |
| HbA1c, mmol/mol | 55 | <48 |
| FSH, IU/L | 7.4 | 1.2–15.8 |
| LH, IU/L | 2.2 | 1.7–8.6 |
ACTH, adrenocorticotropin; FSH, follicle-stimulating hormone; IU, international units; LH, luteinizing hormone; OD, plasma cortisol levels after a 1 mg overnight dexamethasone suppression test; UFC, urine free cortisol hormone.
Differential diagnostic tests
The patient was referred to a tertiary center for further examinations. Ketoconazole treatment was started to alleviate the consequences of hypercortisolism. A pituitary MRI revealed an intrasellar microtumor with a maximal diameter of 6 mm and an IPSS was ordered. A whole-body somatostatin receptor scintigraphy (68Ga-DOTATOC PET/CT) was also performed to evaluate the presence of a potential neuroendocrine tumor. This revealed multiple areas of enhanced uptake including the right thyroid lobe and cervical lymph nodes in the neck (with CT correlates), as well as in the duodenum (with no CT correlate). Concomitantly, a grossly elevated serum calcitonin level of 528 pmol/L (reference range <2.79 pmol/L) was measured.
Subsequently, the IPSS revealed pituitary ACTH secretion with a central-to-peripheral ACTH ratio >3 (Table 2). The right petrosal sinus was not successfully catheterized; thus, lateralization could not be determined.
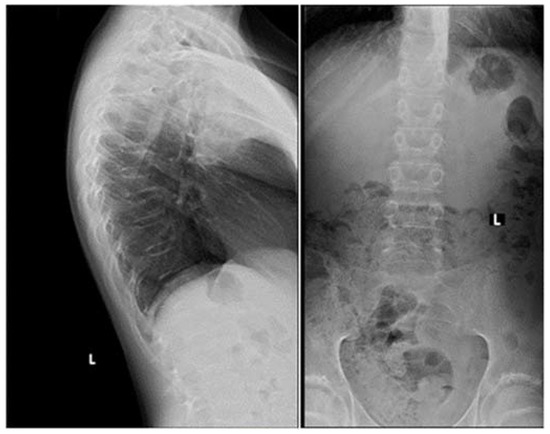
To corroborate the diagnosis MTC, a 18F-DOPA PET/CT scan (FDOPA) was performed (5), which showed pathologically enhanced uptake in the right thyroid lobe and regional lymph nodes (Fig. 1). An ultrasound-guided core needle biopsy from the thyroid nodule was inconclusive; however, the patient underwent total thyroidectomy and regional lymph node resection, from which histology confirmed the diagnosis of disseminated MTC. Standard replacement with levothyroxine, calcium, and vitamin D was initiated. A blood sample was collected, and genomic DNA was extracted. The DNA analysis for RET germline mutation was negative.
 View Full Size
View Full SizeCitation: Endocrinology, Diabetes & Metabolism Case Reports 2023, 3; 10.1530/EDM-23-0057
| Time (min) | Left IPSS | Peripheral | L/P |
|---|---|---|---|
| -5 | 42 | 36 | 1.2 |
| -1 | 116 | 33 | 3.5 |
| 2 | 120 | 32 | 3.8 |
| 5 | 209 | 28 | 7.5 |
| 7 | 180 | 43 | 4.2 |
| 10 | 529 | 34 | 15.6 |
| 15 | 431 | 37 | 11.6 |
*Data represents ACTH levels in ng/L. IPSS Inferior petrosal sampling ACTH Adrenocorticotropin hormone CRH Corticotropin-releasing hormone, L/P Ratio of left (L) inferior petrosal sinus to peripheral venous ACTH concentrations.
Pathology
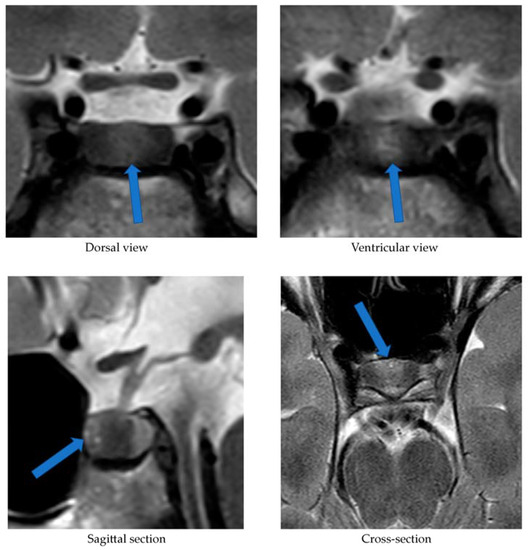
Total thyroidectomy and bilateral cervical lymph node dissection (level six and seven) were performed. Macroscopic evaluation of the right thyroid lobe revealed a 24 mm, irregular solid yellow tumor. Microscopically the tumor showed an infiltrating architecture with pseudofollicles and confluent solid areas. Calcification was prominent, but no amyloid deposition was seen. The tumor cells were pleomorphic with irregular nuclei and heterogenic chromatin structure. No mitotic activity or necrosis was observed. On immunohistochemical analysis, the tumor cells expressed thyroid transcription factor 1 and stained strongly for carcinoembryonic antigen and calcitonin; tumor cells were focally positive for cytokeratin 19. The tumor was completely negative for ACTH, thyroid peroxidase, and the Hector Battifora mesothelial-1 antigen. Further analysis revealed positive immunostaining for CRH (Fig. 2). The Ki-67 index was very low (0–1%), indicating a low cellular proliferation. Molecular testing for somatic RET mutation was not performed.
 View Full Size
View Full SizeCitation: Endocrinology, Diabetes & Metabolism Case Reports 2023, 3; 10.1530/EDM-23-0057
No malignancy was found in the left thyroid lobe and there was no evidence of C-cell hyperplasia. Regional lymph node metastasis was found in 13 out of 15 nodes with extranodal extension.
Outcome and follow-up
Follow-up
Serum calcitonin levels declined after neck surgery but remained grossly elevated (118 pmol/L 3 weeks post surgery) and cortisol levels remained high. Ketoconazole treatment was poorly tolerated and not sufficiently effective. Plasma levels of CRH were measured by a competitive-ELISA kit (EKX-KIZI6R-96 Nordic BioSite), according to the instructions provided by the manufacturer. The intra- and interassay %CV (coefficient of variability) were below 8% and 10%, respectively, and the assay sensitivity was 1.4 pg/mL. The plasma CRH was distinctly elevated compared to in-house healthy controls both before and after thyroid surgery (Fig. 3).
 View Full Size
View Full SizeCitation: Endocrinology, Diabetes & Metabolism Case Reports 2023, 3; 10.1530/EDM-23-0057
The patient subsequently underwent uneventful bilateral laparoscopic adrenalectomy followed by standard replacement therapy with hydrocortisone and fludrocortisone. The symptoms and signs of his CS gradually subsided. Pathology revealed bilateral cortical hyperplasia as expected.
The patient continues follow-up at the Department of Oncology and the Department of Endocrinology and Internal Medicine. At 13-month follow-up, 68Ga-DOTATOC shows residual disease with pathologically enhanced uptake in two lymph nodes, whereas the previously described focal DOTATOC uptake in the duodenum was less pronounced (still no CT correlate). Serum calcitonin was 93 pmol/L at the 13-month follow-up.
Discussion
Diagnostic challenges remain in the distinction between pituitary and ectopic ACTH-dependent CS, and several diagnostic tools are used in combination, none of which is infallible, including IPSS (6). Our case and others illustrate that ectopic CRH secretion may yield a false positive IPSS test result (3). Measurement of circulating CRH levels is relevant if an ectopic CRH producing tumor is suspected, but the assay is not routinely available in clinical practice (Lynnette K Nieman M. Measurement of ACTH, CRH, and other hypothalamic and pituitary peptides https://www.uptodate.com/contents/measurement-of-acth-crh-and-other-hypothalamic-and-pituitary-peptides: UpToDate; 2019). In our case, the presence of elevated plasma CRH levels after thyroidectomy strengthened the indication for bilateral adrenalectomy.
The most common neoplasm causing ectopic CS is small-cell lung cancer, whereas MTC accounts for 2–8% of ectopic cases (7). The development of CS in relation to MTC is generally associated with advanced disease and poor prognosis of an otherwise relatively indolent cancer (8), and the clinical progression of CS is usually rapid, why an early recognition and rapid control of hypercortisolemia and MTC is necessary to decrease morbidity and mortality (7, 9). Our case does have residual disease; however, he remains progression-free with stable and relatively low calcitonin levels within 1-year follow-up.
Only a very limited number of cases of ectopic tumors with either combined ACTH and CRH secretion or isolated CRH secretion have been reported, with ectopic CRH secretion accounting for less than 1% of CS (9).
An ACTH- or CRH-producing tumor can be difficult to localize and may include gastric ACTH/CRH-secreting neuroendocrine tumors (9). In our case, the 68Ga-DOTATOC identified a possible duodenal site, in addition to the MTC, but an upper gastrointestinal endoscopy revealed no pathological findings and there was no CT correlate. Thus, we concluded that the most likely and sole source of ectopic CRH was the MTC and its metastases.
To our knowledge, no official guidelines for managing ectopic ACTH-dependent CS have been established. In a recent publication by Alba et al. (10), the authors demonstrated a clinical algorithm (The Mount Sinai Clinical Pathway, MSCP) and recommendation for the management of CS due to ectopic ACTH secretion.
Essentially, our approach in this particular case followed these recommendations, including source location by CT and 68Ga-DOTATATE PET/CT imaging, acute management with ketoconazole, and finally, bilateral adrenalectomy as curative MTC surgery was not possible. In retrospect, performance of the IPSS could be questioned in view of the MTC diagnosis. In real time, however, a pituitary MRI performed early in the diagnostic process revealed a microadenoma, which prompted the IPSS. In parallel, a somatostatin receptor scintigraphy (68Ga-DOTATOC PET/CT) was also done, which raised the suspicion of an MTC.
Conclusion
We report a very rare case of an ectopic CS caused by a CRH-secreting MTC. Although IPSS has stood the test of time in the differential diagnosis of ACTH-dependent CS, this case illustrates a rare pitfall.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial, or not-for-profit sector.
Patient consent
Written informed consent for publication of their clinical details was obtained from the patient.
Author contribution statement
JOJ and MJO are the physicians responsible for the patient. LR performed the thyroidectomy and bilateral adrenalectomy. SHM and SLA assessed and reassessed the histopathology and the immunohistochemical analysis. MB measured plasma CRH. VM, JOJ, and MJO drafted the manuscript. All authors contributed to critical revision of the manuscript.
References
-
1↑Raff H, & Carroll T. Cushing’s syndrome: from physiological principles to diagnosis and clinical care. Journal of Physiology 2015 593 493–506. (https://doi.org/10.1113/jphysiol.2014.282871)
-
2↑Hatipoglu BA. Cushing’s syndrome. Journal of Surgical Oncology 2012 106 565–571. (https://doi.org/10.1002/jso.23197)
-
3↑Vassiliadi DA, Mourelatos P, Kratimenos T, & Tsagarakis S. Inferior petrosal sinus sampling in Cushing’s syndrome: usefulness and pitfalls. Endocrine 2021 73 530–539. (https://doi.org/10.1007/s12020-021-02764-4)
-
4↑Zampetti B, Grossrubatscher E, Dalino Ciaramella P, Boccardi E, & Loli P. Bilateral inferior petrosal sinus sampling. Endocrine Connections 2016 5 R12–R25. (https://doi.org/10.1530/EC-16-0029)
-
5↑Treglia G, Rufini V, Salvatori M, Giordano A, & Giovanella L. PET imaging in recurrent medullary thyroid carcinoma. International Journal of Molecular Imaging 2012 2012 324686. (https://doi.org/10.1155/2012/324686)
-
6↑Fasshauer M, Lincke T, Witzigmann H, Kluge R, Tannapfel A, Moche M, Buchfelder M, Petersenn S, Kratzsch J, Paschke R, et al.Ectopic Cushing’ syndrome caused by a neuroendocrine carcinoma of the mesentery. BMC Cancer 2006 6 108. (https://doi.org/10.1186/1471-2407-6-108)
-
7↑Chrisoulidou A, Pazaitou-Panayiotou K, Georgiou E, Boudina M, Kontogeorgos G, Iakovou I, Efstratiou I, Patakiouta F, & Vainas I. Ectopic Cushing’s syndrome due to CRH secreting liver metastasis in a patient with medullary thyroid carcinoma. Hormones 2008 7 259–262. (https://doi.org/10.1007/BF03401514)
-
8↑Corsello A, Ramunno V, Locantore P, Pacini G, Rossi ED, Torino F, Pontecorvi A, De Crea C, Paragliola RM, Raffaelli M, et al.Medullary thyroid cancer with ectopic Cushing’s syndrome: a case report and systematic review of detailed cases from the literature. Thyroid 2022 32 1281–1298. (https://doi.org/10.1089/thy.2021.0696)
-
9↑Sharma ST, Nieman LK, & Feelders RA. Cushing’s syndrome: epidemiology and developments in disease management. Clinical Epidemiology 2015 7 281–293. (https://doi.org/10.2147/CLEP.S44336)
-
10↑Alba EL, Japp EA, Fernandez-Ranvier G, Badani K, Wilck E, Ghesani M, Wolf A, Wolin EM, Corbett V, Steinmetz D, et al.The Mount Sinai clinical pathway for the diagnosis and management of hypercortisolism due to ectopic ACTH syndrome. Journal of the Endocrine Society 2022 6 bvac073. (https://doi.org/10.1210/jendso/bvac073)
From https://edm.bioscientifica.com/view/journals/edm/2023/3/EDM23-0057.xml
Filed under: Cancer, Cushing's, Diagnostic Testing, Rare Diseases, symptoms | Tagged: corticotropin-releasing hormone, ectopic, Inferior petrosal sinus sampling, IPSS, male, medullary thyroid carcinoma | Leave a comment »