Patients who are diagnosed with pituitary disease experience many physical changes that have been well documented in the literature. These patients may have significant fluctuations in their weight as well as changes in their physical appearance, as seen in patients with adrenocorticotropic hormone-producing adenomas (ACTHomas), prolactinomas, and growth hormone-producing adenomas (GHomas). They may also experience changes in their sexual and reproductive functioning, such as amenorrhea, impotence, and impaired orgasm. Many patients with pituitary disease develop comorbid medical illnesses as a result of their pituitary dysfunction. The development of diabetes mellitus, hypertension, and coronary artery disease are a few examples.
In addition to the physical changes and comorbid medical conditions, there is a clear association between psychiatric illness and pituitary disease. In pituitary disease, one of the most common psychiatric manifestations is the presence of depressive symptoms significant enough to warrant diagnosis of a major depressive disorder.1 Cushing’s disease is probably the most frequently studied of the pituitary diseases. It has been shown that roughly 50% of patients with Cushing’s disease have depressive symptoms of a sufficient intensity to be diagnosed with a major depressive disorder. In addition to the depressive symptoms, these patients often demonstrate pronounced psychopathology associated with the depression, including lethargy, increased sleepiness, cyclic mood instability, impaired cognitive function (i.e., deficits in concentration and memory), and personality change.2,3 Similarly, patients with growth hormone deficiency (GHD) may experience a lack of energy and drive (i.e., motivation), as well as problems with memory and concentration.4
Patients with acromegaly have also been shown to have psychiatric disturbances. Up to 75% of patients experience personality changes, including depressive symptoms, other mood changes, anger, loss of motivation, and a decreased willingness to socialize.5 Patients with prolactinomas also have reported a higher frequency of depressive symptoms, anxiety, and feelings of hostility than a group of people with either amenorrhea associated with normal prolactin levels or healthy comparison subjects with normal prolactin levels.6 In addition, apathy was one of four neuropsychiatric signs and symptoms (i.e., asexuality, adiposity, headache).7
Few studies have attempted to quantify the overall impact that a variety of pituitary diseases has on social functioning and overall quality of life. One study reported that patients with adult-onset pituitary insufficiency experienced decreased quality of life. The patients who had adult-onset GHD experienced a lower sense of well-being and a decreased psychological reserve (i.e., coping reserve) than a reference population. They also had lower scores on measures of perceived health and social function than the reference population.4
Another study compared pituitary tumor patients based on treatments received (i.e., surgery either transfrontally or transsphenoidally and medical management) to a group of comparison subjects.8 The results showed that the patient groups who had received either transsphenoidal surgery or medical management rated themselves as having mild mood disturbance, poorer social adjustment, higher levels of emotional distress, and lower energy levels than comparison subjects. Although the transfrontal surgery patients rated themselves no differently than the comparison group, proxy assessment revealed these patients to be different from the comparison group as well. The investigators concluded that this discrepancy was related to the lack of insight associated with frontal lobe surgery and damage.
Pituitary patients who received radiotherapy have been shown to have lower quality of life scores, when compared to healthy subjects.9 Furthermore, in a study that compared the performance of pituitary tumor patients with and without radiotherapy, both pituitary tumor groups performed poorer than the comparison group, with similar degrees of dysfunction in executive functioning, verbal memory and visual memory for both tumor groups.10
Thus, given the paucity of research, studies across all pituitary diagnoses document the presence of depressive symptoms, mood instability, loss of motivation, and anxiety. It also appears that, when matched against comparison groups, these patients experience lower quality of life and more cognitive dysfunction if they receive radiotherapy. Clearly, there are limitations to the work that has been described. These studies have all involved small sample sizes, so a question of adequate power to detect meaningful differences between the groups comes into question. In addition, the majority of these studies has focused on all pituitary patients, and it appears that issues may be different for different pituitary diagnoses. Finally, the majority of these studies has not controlled for other factors that may interfere with interpretation of the results, such as comorbid medical illness. As a result, it is difficult to generalize the results regarding neurocognitive functioning.
Furthermore, the studies to date have focused on the presence of depressive symptomatology, as measured by self-report instruments. Thus, depression is not being defined on a syndromal level, as it would be if a structured diagnostic interview was used. Without the use of a structured diagnostic interview for depression, it would be difficult to separate clinical depression from a secondary mood disorder directly related to the hormonal effects of the pituitary tumor. Conclusions about concentration and memory difficulties, as well as lack of motivation, are also being made using patient self-report as opposed to neuropsychological assessment (Table 1).
The fact remains, however, that these patients self-report depressive symptoms, lack of motivation, and cognitive impairment. The question that emerges is whether the symptoms and concerns that appear to reflect major depression, chronic fatigue, or apathy are caused by the syndrome of major depression, are sequelae of fatigue or apathy, or are due to some third factor independent of depression, fatigue, or apathy.
Our premise is that patients with pituitary disease have a neurobiological illness with dysfunction in the cerebral hemispheres and the diencephalons that manifests as apathy and fatigue. We present four cases (Appendix) of patients with pituitary disease presenting to Moffitt Cancer Center with depressive symptoms, but who appear to meet criteria for chronic fatigue or apathy syndrome. In this article we will discuss chronic fatigue and apathy syndromes and their relationship to pituitary disease, as well as potential treatments for chronic fatigue and apathy syndrome that are applicable to pituitary disease.
DISCUSSION
Section:
Choose
Top of page
Abstract
CASE SERIES
DISCUSSION <<
References
CITING ARTICLES
These four cases illustrate an example of a neurobiological illness in which patients report symptoms that appear to be part of the syndrome of depression but, in fact, are not. Instead, these symptoms encompass an overlapping problem with clinical depression, namely fatigue and apathy, which is very responsive to stimulant medications.
Fatigue has been defined as a clinical syndrome characterized with generalized weakness, and there is also an interrelationship between physical, emotional, and mental fatigue.11 Physical fatigue is a state in which individuals experience a sustained sense of exhaustion and decreased capacity for physical and mental work that can be restored with rest and support.12 The emotional effects of fatigue are well known in psychiatry. Numerous psychiatric illnesses, including major depressive disorder, generalized anxiety disorder, and dysthymia are frequently associated with fatigue. In fact, more than two thirds of patients with clinical depression present with signs of fatigue that may include low energy and listlessness.13 It has been proposed that the syndromes of fatigue and depression may represent overlapping constructs, sharing similar underlying mechanisms,13–15 a hypothesis that is supported by observations that patients with clinical depression, who respond to antidepressant medication, may continue to experience residual fatigue.16
Fatigue, as a mental state, displays a complex interaction of subjective feelings, tissue impairment and diminished performance.17 In fact, chronic fatigue, such as that observed in chronic fatigue syndrome, shares a number of cardinal symptoms with major depression, including profound fatigue; decreased, nonrestorative sleep; and decreased concentration and memory.18 However, these syndromes are associated with significantly different physiological abnormalities.19,20 In addition, unlike depressed patients, patients with chronic fatigue do not seem to respond to antidepressant medications.21,22 Such was the case for the subjects described in this report.
Furthermore, it has been shown that chronic fatigue patients consistently report lower mean scores on depression inventories than depressed patients, although their scores were still within the depressed range.23 Edwards et al.23 also showed that depressed patients scored significantly higher on the self-reproach or cognitive symptoms, including feelings of guilt, low self-esteem, and suicidal ideation, accounting for the differences in the groups. Another study expanded on these findings, comparing clinically depressed patients, depressed patients with chronic fatigue syndrome, nondepressed patients with chronic fatigue syndrome, and healthy comparison subjects.18 The clinically depressed group had significantly lower self-esteem, increased cognitive distortions across all situations, and was more likely to attribute their illness to psychological factors. The chronic fatigue patients rated their current health status lower, had a strong illness identity, and attributed their illness to external factors. Their distortions in thinking were specific to somatic experiences and they were more likely than depressed patients to cope with their illness by limiting stress and activity levels. The subjects in our report also stated specifically that their emotional and cognitive symptoms were attributable to their underlying medical illness and that their medical illness was something that was outside of their control.
The above observations suggest that what chronic fatigue patients, including our patients with pituitary disease, experience is not depression. However, what these patients experience also needs to be differentiated from demoralization, another psychological state. Demoralization is experienced as a persistent inability to cope, together with associated feelings of helplessness, hopelessness, meaninglessness, subjective incompetence, and diminished self-esteem.24 When coping is insufficient and the person is at a point when he or she does not know how to proceed, distress and helplessness result. While clinical depression is characterized primarily by anhedonia,25,26 demoralization is characterized by a feeling of subjective incompetence and helplessness. Any number of events can threaten a person’s sense of independence and competence, including both physical and mental illness.27 Chronic illness can threaten the integrity of the body and mind, challenging a person’s mastery and control. Patients often need to reduce social roles, depriving them of a sense of satisfaction and competence. With reduction of personal efficacy, and uncertain prognosis, comes a sense of demoralization.28,29 Our patients did not feel helpless or incompetent. In fact, they continued to actively seek help from different physicians because they knew that there was something wrong with their bodies that no one was addressing.
While fatigue affects physical, emotional, and mental areas of functioning, it also affects cognitive function by affecting the ability to focus or pay attention. The ability to focus and concentrate is essential for effective functioning in daily life.30 Directed attention is the capacity to inhibit or block competing stimuli or distractions during purposeful activity. Directed attention is what helps us function in goal-directed activities and regulates behavior to meet desired goals. Intense or prolonged demands on attentional capacity can lead to a decline in the ability to maintain directed attention, thus causing attentional fatigue. In healthy adults attentional fatigue has been associated with impaired ability to perceive, interpret and respond to environmental information and distressing affective states.31
The construct of fatigue, as described above, shares many similarities with the concept of apathy. There are three components to apathy: observable behavior, emotional content, and thought content.2,32 As in fatigue, all three components are affected simultaneously. It is not simply the lack of emotion that defines apathy; it is the diminished emotional responsiveness to goal-related events and decreased emotional distress that define apathy.2,32 Recent neurophysiological and neurocognitive research have provided a greater appreciation of the functional neuroanatomy of fatigue and apathy.33–35 Regarding fatigue, the frontal lobes, posterior parietal lobes, and the reticular formation are primarily involved in attention control. The posterior parietal cortex is associated with shift in attention focus from one spatial location to another and from one stimulus to another.36 The frontal lobes are associated with controlling selection of stimuli from competing inputs and exercising inhibitory control over other brain areas to perform complex tasks. In addition, the subcortical system, including the reticular formation in the brain stem, is responsible for alertness and waking states.36
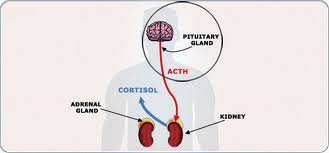
Other areas that may play a role in fatigue and apathy include the medial dorsal nucleus of the thalamus, caudate nucleus, nucleus accumbens and globus pallidus.2 Thus, the circuit most implicated in this syndrome is the cortical-striatal-thalamic-cortical circuit. This circuit helps elucidate the mechanism behind fatigue and apathy, in general. How does this pertain to patients with pituitary disease? The hypothalamic-pituitary tract provides significant afferent input to the mediodorsal nucleus of the thalamus and any perturbation in hypothalamic-pituitary function will affect the frontal-subcortical circuits and cause apathy syndrome.2 However, when thinking of pituitary tumors and psychiatric disturbances, the deficits are related to the direct and indirect effects of the tumor and the hormonal alterations caused by the tumor. The neuropsychological impairment caused by these tumors has been documented to cause problems with executive functioning, verbal reasoning, and visual memory.2
In apathy syndrome it is believed that mesocortical tract hypoactivity causes impaired executive functioning and that mesolimbic tract hypoactivity is responsible for the affective component of apathy.2 Norepinephrine systems in the brain have been linked to attentional operations. Medications that increase noradrenergic activity enhance concentration, and disturbing this system has been shown to interfere with attention.36 The functioning of the frontal lobes and norepinephrine’s effect on attention constitute the primary relationship between fatigue and apathy syndrome.
Methylphenidate is a medication well known for its use in the treatment of Attention Deficit Hyperactivity Disorder (ADHD)/Attention Deficit Disorder (ADD).37 Preclinical studies have shown that stimulants, including methylphenidate, block the reuptake of dopamine and norepinephrine into the presynaptic neurons and increase the release of these neurotransmitters into the extraneuronal space.38 In ADD, a hypoperfusion of the striatum and hyperperfusion to the primary sensory regions were demonstrated by Xenon inhalation techniques.39 However, after the addition of methylphenidate, the striatal and posterior periventricular regions had increased blood flow. Conversely, after the addition of methylphenidate the primary sensory regions showed decreased blood flow. Additionally, after administration of methylphenidate PET scans have shown an increase in blood flow to the mesencephalon and basal ganglia, and conversely, motoric cortical areas have shown a decrease in blood flow.39 These changes mentioned above could provide the mechanism for improvement in children with ADHD taking methylphenidate.
The patients in our case series meet the criteria of apathy syndrome because of their impaired cognitive and affective functioning. As mentioned earlier, pituitary tumors can cause apathy syndrome by their influence on frontal-subcortical pathways. In our patients, we observed improvement after they started taking methylphenidate. Subjective improvements were described as increased ability to focus, increased memory, increased functioning at work, improved confidence, and improved mental stamina. These patients showed improvements in: new learning of verbal and nonverbal information; sustained effortful output as assessed with a verbal fluency task; executive function across tasks tapping novel problem solving; the ability to inhibit responding, sequencing, and mental flexibility; and psychomotor speed. Through intervention with methylphenidate the patients appeared to show significant improvement subjectively and objectively.
Many physicians have used stimulants to help with patients suffering from brain tumors and cognitive slowing. One case study suggested that methylphenidate improved severe neurobehavioral slowing caused by frontal and brain stem dysfunction due to cancer and cancer treatment.40 The patients in this study showed improvement in arousal, attention, and speed on initiation of tasks and were able to concentrate on tasks longer. Another study tested the effect of methylphenidate on patients with primary gliomas who had neurobehavioral slowing affecting their level of functioning.41 These patients showed an improvement in cognitive and daily functioning while taking methylphenidate. They also showed improvement in visual motor speed, verbal memory, expansive speech, and executive functioning, as well as improvement in cognition and mood.41 It is important to review, with the patient, the risks and benefits of treatment with methylphenidate, determining that the patient does not have uncontrolled hypertension, recent myocardial infarction, or glaucoma. A review of the potential for abuse, while extremely low as compared to the amphetamines, is also necessary.
In summary, apathy is an emerging concept in neuropsychiatry, and major depressive disorder and chronic fatigue syndrome are important syndromes from which apathy must be differentiated. As was demonstrated by our series of cases, these patients with pituitary disease appeared to be suffering from depression, given their constricted affect. However, when asked about their mood, all stated that they were not depressed but, instead, stated that they had chronic fatigue and lack of motivation. All responded well to methylphenidate and not to antidepressants. As more and more patients with pituitary disease present to cancer centers for evaluation and treatment, it is incumbent on the psychosocial staff to closely scrutinize the pituitary patient for mood changes. Since many of these patients will appear depressed, yet deny depressed mood and anhedonia, it is important to keep apathy syndrome in mind, particularly when these patients do not respond or their depression appears refractory to traditional antidepressants. Apathy syndrome is easily treated with stimulant medications, leading to increased energy and mood.










 Radiation therapy can be effective in controlling the growth of the tumor. However, if you received radiation therapy in the past, additional radiation may not be safe.
Radiation therapy can be effective in controlling the growth of the tumor. However, if you received radiation therapy in the past, additional radiation may not be safe. Medical therapies for the treatment of Nelson’s syndrome are currently limited, but include:
Medical therapies for the treatment of Nelson’s syndrome are currently limited, but include: