Cushing’s syndrome (CS) arises from an excess of endogenous or exogenous cortisol, with Cushing’s disease specifically implicating a pituitary adenoma and exaggerated adrenocorticotropic hormone (ACTH) production. Typically, Cushing’s disease presents with characteristic symptoms such as weight gain, central obesity, moon face, and buffalo hump.
This case report presents an unusual manifestation of CS in a 48-year-old male with a history of hypertension, where severe hypokalemia was the primary presentation. Initial complaints included bilateral leg swelling, muscle weakness, occasional shortness of breath, and a general feeling of not feeling well. Subsequent investigations revealed hypokalemia, metabolic alkalosis, and an abnormal response to dexamethasone suppression, raising concerns about hypercortisolism. Further tests, including 24-hour urinary free cortisol and ACTH testing, confirmed significant elevations. Brain magnetic resonance imaging (MRI) identified a pituitary macroadenoma, necessitating neurosurgical intervention.
This case underscores the rarity of CS presenting with severe hypokalemia, highlighting the diagnostic challenges and the crucial role of a collaborative approach in managing such intricate cases.
Introduction
Cushing’s syndrome (CS), characterized by excessive cortisol production, is well-known for its diverse and often conspicuous clinical manifestations. Cushing’s disease is a subset of CS resulting from a pituitary adenoma overproducing adrenocorticotropic hormone (ACTH), leading to heightened cortisol secretion. The classic presentation involves a spectrum of symptoms such as weight gain, central obesity, muscle weakness, and mood alterations [1].
Despite its classic presentation, CS can demonstrate diverse and atypical features, challenging conventional diagnostic paradigms. This case report sheds light on a rare manifestation of CS, where severe hypokalemia was the primary clinical indicator. Notably, instances of CS prominently manifesting through severe hypokalemia are scarce in the literature [1,2].
Through this exploration, we aim to provide valuable insights into the diagnostic intricacies of atypical CS presentations, underscore the significance of a comprehensive workup, and emphasize the collaborative approach essential for managing such uncommon hormonal disorders.
Case Presentation
A 48-year-old male with a history of hypertension presented to his primary care physician with complaints of bilateral leg swelling, occasional shortness of breath, dizziness, and a general feeling of malaise persisting for 10 days. The patient reported increased water intake and urinary frequency without dysuria. The patient was diagnosed with hypertension eight months ago. He experienced progressive muscle weakness over two months, hindering his ability to perform daily activities, including using the bathroom. The primary care physician initiated a blood workup that revealed severe hypokalemia with a potassium level of 1.3 mmol/L (reference range: 3.6 to 5.2 mmol/L), prompting referral to the hospital.
Upon admission, the patient was hypertensive with a blood pressure of 180/103 mmHg, a heart rate of 71 beats/minute, a respiratory rate of 18 breaths/minute, and an oxygen saturation of 96% on room air. Physical examination revealed fine tremors, bilateral 2+ pitting edema in the lower extremities up to mid-shin, abdominal distension with normal bowel sounds, and bilateral reduced air entry in the bases of the lungs on auscultation. The blood work showed the following findings (Table 1).
Parameter
Result
Reference Range
Potassium (K)
1.8 mmol/L
3.5-5.0 mmol/L
Sodium (Na)
144 mmol/L
135-145 mmol/L
Magnesium (Mg)
1.3 mg/dL
1.7-2.2 mg/dL
Hemoglobin (Hb)
15.5 g/dL
13.8-17.2 g/dL
White blood cell count (WBC)
13,000 x 103/µL
4.5 to 11.0 × 109/L
Platelets
131,000 x 109/L
150-450 x 109/L
pH
7.57
7.35-7.45
Bicarbonate (HCO3)
46 mmol/L
22-26 mmol/L
Lactic acid
4.2 mmol/L
0.5-2.0 mmol/L
Table 1: Blood work findings
In order to correct the electrolyte imbalances, the patient received intravenous (IV) magnesium and potassium replacement and was later transitioned to oral. The patient was also started on normal saline at 100 cc per hour. To further investigate the complaint of shortness of breath, the patient underwent a chest X-ray, which revealed bilateral multilobar pneumonia (Figure 1). He was subsequently treated with ceftriaxone (1 g IV daily) and clarithromycin (500 mg twice daily) for seven days.
Figure 1: A chest X-ray revealing (arrows) bilateral multilobar pneumonia
With persistent abdominal pain and lactic acidosis, a computed tomography (CT) scan abdomen and pelvis with contrast was conducted, revealing a psoas muscle hematoma. Subsequent magnetic resonance imaging (MRI) depicted an 8×8 cm hematoma involving the left psoas and iliacus muscles. The interventional radiologist performed drainage of the hematoma involving the left psoas and iliacus muscles (Figure 2).
Figure 2: Magnetic resonance imaging (MRI) depicting an 8×8 cm hematoma (arrow) involving the left psoas and iliacus muscles
In light of the concurrent presence of hypokalemia, hypertension, and metabolic alkalosis, there arose concerns about Conn’s syndrome, prompting consultation with endocrinology. Their recommended workup for Conn’s syndrome included assessments of the aldosterone-renin ratio and random cortisol levels. The results unveiled an aldosterone level below 60 pmol/L (reference range: 190 to 830 pmol/L in SI units) and a plasma renin level of 0.2 pmol/L (reference range: 0.7 to 3.3 mcg/L/hr in SI units). Notably, the aldosterone-renin ratio was low, conclusively ruling out Conn’s syndrome. The random cortisol level was notably elevated at 1334 nmol/L (reference range: 140 to 690 nmol/L).
Furthermore, a low-dose dexamethasone suppression test was undertaken due to the high cortisol levels. Following the administration of 1 mg of dexamethasone at 10 p.m., cortisol levels were measured at 9 p.m., 3 a.m., and 9 a.m. the following day. The results unveiled a persistently elevated cortisol level surpassing 1655 nmol/L, signaling an abnormal response to dexamethasone suppression and raising concerns about a hypercortisolism disorder, such as CS.
In the intricate progression of this case, the investigation delved deeper with a 24-hour urinary free cortisol level, revealing a significant elevation at 521 mcg/day (reference range: 10 to 55 mcg/day). Subsequent testing of ACTH portrayed a markedly elevated level of 445 ng/L, distinctly exceeding the normal reference range of 7.2 to 63.3 ng/L. A high-dose 8 mg dexamethasone test was performed to ascertain the source of excess ACTH production. The baseline serum cortisol levels before the high-dose dexamethasone suppression test were 1404 nmol/L, which decreased to 612 nmol/L afterward, strongly suggesting the source of excess ACTH production to be in the pituitary gland.
A CT scan of the adrenal glands ruled out adrenal mass, while an MRI of the brain uncovered a 1.3×1.3×3.2 cm pituitary macroadenoma (Figure 3), leading to compression of adjacent structures. Neurosurgery was consulted, and they recommended surgical removal of the macroadenoma due to the tumor size and potential complications. The patient was referred to a tertiary care hospital for pituitary adenoma removal.
Figure 3: Magnetic resonance imaging (MRI) of the brain depicting a 1.3×1.3×3.2 cm pituitary macroadenoma (star)
Discussion
CS represents a complex endocrine disorder characterized by excessive cortisol production. While the classic presentation of CS includes weight gain, central obesity, and muscle weakness, our case highlights an uncommon initial manifestation: severe hypokalemia. This atypical presentation underscores the diverse clinical spectrum of CS and the challenges it poses in diagnosis and management [1,2].
While CS typically presents with the classic symptoms mentioned above, severe hypokalemia as the initial manifestation is exceedingly rare. Hypokalemia in CS often results from excess cortisol-mediated activation of mineralocorticoid receptors, leading to increased urinary potassium excretion and renal potassium wasting. Additionally, metabolic alkalosis secondary to cortisol excess further exacerbates hypokalemia [3,4].
Diagnosing a case of Cushing’s disease typically commences with a thorough examination of the patient’s medical history and a comprehensive physical assessment aimed at identifying characteristic manifestations such as central obesity, facial rounding, proximal muscle weakness, and increased susceptibility to bruising. Essential to confirming the diagnosis are laboratory examinations, which involve measuring cortisol levels through various tests, including 24-hour urinary free cortisol testing, late-night salivary cortisol testing, and dexamethasone suppression tests. Furthermore, assessing plasma ACTH levels aids in distinguishing between pituitary-dependent and non-pituitary causes of CS. Integral to the diagnostic process are imaging modalities such as MRI of the pituitary gland, which facilitate the visualization of adenomas and the determination of their size and precise location [1-4].
Treatment for Cushing’s disease primarily entails surgical removal of the pituitary adenoma via transsphenoidal surgery, with the aim of excising the tumor and restoring normal pituitary function. In cases where surgical intervention is unsuitable or unsuccessful, pharmacological therapies employing medications such as cabergoline (a dopamine receptor agonist) or pasireotide (a somatostatin analogue) may be considered to suppress ACTH secretion and regulate cortisol levels. Additionally, radiation therapy, whether conventional or stereotactic radiosurgery, serves as a supplementary or alternative treatment approach to reduce tumor dimensions and mitigate ACTH production [5,6]. To assess the effectiveness of treatment, manage any problem, and assure long-term illness remission, diligent long-term follow-up and monitoring are essential. Collaborative multidisciplinary care involving specialists such as endocrinologists, neurosurgeons, and other healthcare professionals is pivotal in optimizing patient outcomes and enhancing overall quality of life [2,4].
The prognosis of CS largely depends on the underlying cause, stage of the disease, and efficacy of treatment. Early recognition and prompt intervention are essential for improving outcomes and minimizing long-term complications. Surgical resection of the adrenal or pituitary tumor can lead to remission of CS in the majority of cases. However, recurrence rates vary depending on factors such as tumor size, invasiveness, and completeness of resection [2,3]. Long-term follow-up with endocrinologists is crucial for monitoring disease recurrence, assessing hormonal function, and managing comorbidities associated with CS.
Conclusions
In conclusion, our case report highlights the rarity of severe hypokalemia as the initial presentation of CS. This unique presentation underscores the diverse clinical manifestations of CS and emphasizes the diagnostic challenges encountered in clinical practice. A multidisciplinary approach involving endocrinologists, neurosurgeons, and radiologists is essential for the timely diagnosis and management of CS. Early recognition, prompt intervention, and long-term follow-up are essential for optimizing outcomes and improving the quality of life for patients with this endocrine disorder.
So, these are only seven of the many, many symptoms of Cushing’s. I had those above – and I often felt like I looked like one of those little bearded dwarves.
Cushing’s affects every part of the body. It’s not like when I had kidney cancer and only the kidney was affected.
Here are some of the many areas affected.
Progressive obesity and skin changes
Weight gain and fatty tissue deposits, particularly around the midsection and upper back, in the face (moon face) and between the shoulders (buffalo hump). Some symptoms such as sudden weight gain, are caused by excess cortisol. The excess cortisol in the body does not increase protein and carbohydrate metabolism. It slows or nearly disables metabolism function, which can cause weight gain (fat accumulation) in the buttocks, abdomen, cheeks, neck, or upper back.
Loss of muscle mass. Some areas of the body, such as the arms and legs, will remain thin.
Pink or purple stretch marks (striae) on the skin of the abdomen, thighs, breasts and arms
Thinning, fragile skin that bruises easily
Slow healing of cuts, insect bites and infections
Acne
Women with Cushing’s syndrome may experience:
Thicker or more visible body and facial hair (hirsutism)
Irregular or absent menstrual periods
Men with Cushing’s syndrome may experience:
Decreased libido
Decreased fertility
Erectile dysfunction
Other signs and symptoms include:
Fatigue
Muscle weakness
Depression, anxiety and irritability
Loss of emotional control
Cognitive difficulties
New or worsened high blood pressure
Glucose intolerance that may lead to diabetes
Headache
Bone loss, leading to fractures over time
Hyperlipidemia (elevated lipids – cholesterol – in the blood stream)
Recurrent opportunistic or bacterial infections
Think you have Cushing’s? Get to a doctor and don’t give up!
The diagnosis of Cushing’s syndrome is challenging; however, through the clinical picture and the search for secondary causes of osteoporosis, it was possible to reach the diagnosis of the case reported. There was an independent, symptomatic ACTH hypercortisolism manifested by typical phenotypic changes, severe secondary osteoporosis and arterial hypertension in a young patient.
Case presentation
A 20-year-old Brazilian man with low back pain for 8 months. Radiographs showed fragility fractures in the thoracolumbar spine, and bone densitometry showed osteoporosis, especially when evaluating the Z Score (− 5.6 in the lumbar spine). On physical examination, there were wide violaceous streaks on the upper limbs and abdomen, plethora and fat increase in the temporal facial region, hump, ecchymosis on limbs, hypotrophy of arms and thighs, central obesity and kyphoscoliosis. His blood pressure was 150 × 90 mmHg. Cortisol after 1 mg of dexamethasone (24.1 µg/dL) and after Liddle 1 (28 µg/dL) were not suppressed, despite normal cortisoluria. Tomography showed bilateral adrenal nodules with more severe characteristics. Unfortunately, through the catheterization of adrenal veins, it was not possible to differentiate the nodules due to the achievement of cortisol levels that exceeded the upper limit of the dilution method. Among the hypotheses for the differential diagnosis of bilateral adrenal hyperplasia are primary bilateral macronodular adrenal hyperplasia, McCune–Albright syndrome and isolated bilateral primary pigmented nodular hyperplasia or associated with Carney’s complex. In this case, primary pigmented nodular hyperplasia or carcinoma became important etiological hypotheses when comparing the epidemiology in a young man and the clinical-laboratory-imaging findings of the differential diagnoses. After 6 months of drug inhibition of steroidogenesis, blood pressure control and anti-osteoporotic therapy, the levels and deleterious metabolic effects of hypercortisolism, which could also impair adrenalectomy in the short and long term, were reduced. Left adrenalectomy was chosen, given the possibility of malignancy in a young patient and to avoid unnecessary definitive surgical adrenal insufficiency if the adrenalectomy was bilateral. Anatomopathology of the left gland revealed expansion of the zona fasciculate with multiple nonencapsulated nodules.
Conclusion
The early identification of Cushing’s syndrome, with measures based on the assessment of risks and benefits, remains the best way to prevent its progression and reduce the morbidity of the condition. Despite the unavailability of genetic analysis for a precise etiological definition, it is possible to take efficient measures to avoid future damage.
Cushing’s syndrome may be exogenous or endogenous and, in this case, can be ACTH-dependent or independent. In the case reported, there was an independent, symptomatic ACTH hypercortisolism manifested by typical phenotypic changes, severe secondary osteoporosis and arterial hypertension in a young patient. Osteoporosis secondary to hypercortisolism occurs due to chronic reduction in bone formation, loss of osteocytes and increased reabsorption caused by intense binding of cortisol to glucocorticoid receptors present in bone cells [1]. In addition, excess cortisol impairs vitamin D metabolism and reduces endogenous parathyroid hormone secretion, intestinal calcium reabsorption, growth hormone release, and lean body mass [2]. Subclinical Cushing disease occurs in up to 11% of individuals diagnosed with early-onset osteoporosis and 0.5–1% of hypertension patients. [3] A cross-sectional study published in 2023 revealed a prevalence of 81.5% bone loss in 19 patients with Cushing’s syndrome [2]. The prevalence of osteopenia ranges from 60 to 80%, and the prevalence of osteoporosis ranges from 30 to 65% in patients with Cushing’s syndrome. Additionally, the incidence of fragility fractures ranges from 30 to 50% in these patients [4] and is considered the main cause of morbidity affecting the quality of life. The diagnosis is challenging, given the presence of confounding factors; however, through the clinical picture and the search for secondary causes of osteoporosis, it was possible to reach a syndromic diagnosis. Early identification of this syndrome, with measures based on the assessment of risks and benefits, remains the best way to prevent progression and reduce morbidity related to this disease [2].
Case presentation
A 20-year-old Brazilian male patient reported low back pain that had evolved for 8 months, with no related trauma. He sought emergency care and performed spinal radiographs on this occasion (03/2019). Due to the several alterations observed in the images, he was referred to the Orthopedics Service of the Hospital of Federal University of Juiz de Fora, which prescribed orthopedic braces, indicated physical therapy and was referred again to the Osteometabolic Diseases outpatient clinic of the Endocrinology and Rheumatology Services of the Hospital of Federal University of Juiz de Fora on 10/2019.
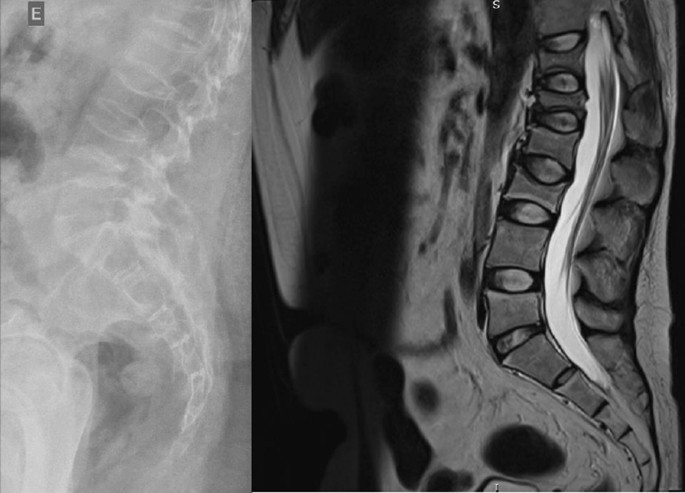
The radiographs showed a marked reduction in the density of bone structures, scoliotic deviation with convexity toward the left and reduction in the height of the lumbar vertebrae, with partial collapses of the vertebral bodies at the level of T12, L1, L2, L3 and L5, with recent collapses in T12 and L1, suggesting bone fragility fractures. The same can be seen in posterior magnetic resonance imaging (Fig. 1).
Fig. 1
Radiography and Magnetic Resonance Imaging (MRI) of lumbosacral spine in profile
Bone scintigraphy on 08/2019 did not reveal hyper flow or anomalous hyperemia in the topography of the thoracolumbar spine, and in the later images of the exam, there was a greater relative uptake of the tracer in the lumbar spine (vertebrae T10–T12, L2–L4), of nonspecific aspect, questioning the presence of osteoarticular processes or ankylosing spondylitis.
It was also observed in the bone densitometry requested in October 2019, performed by dual-energy X-ray absorptiometry (DXA), low bone mineral density (BMD) in the lumbar spine, femoral neck and total femur, when comparing the results to evaluating the Z Score (Table 1).
Thus, the diagnosis of osteoporosis was established, and treatment with vitamin D 7000 IU per week was started due to vitamin D3 insufficiency associated with the bisphosphonate alendronate 70 mg, also weekly. The patient had a past pathological history of fully treated syphilis (2018) and perianal condyloma with a surgical resection on 09/2017 and 02/2018. In the family history, it was reported that a maternal uncle died of systemic sclerosis. In the social context, the young person denied drinking alcohol and previous or current smoking.
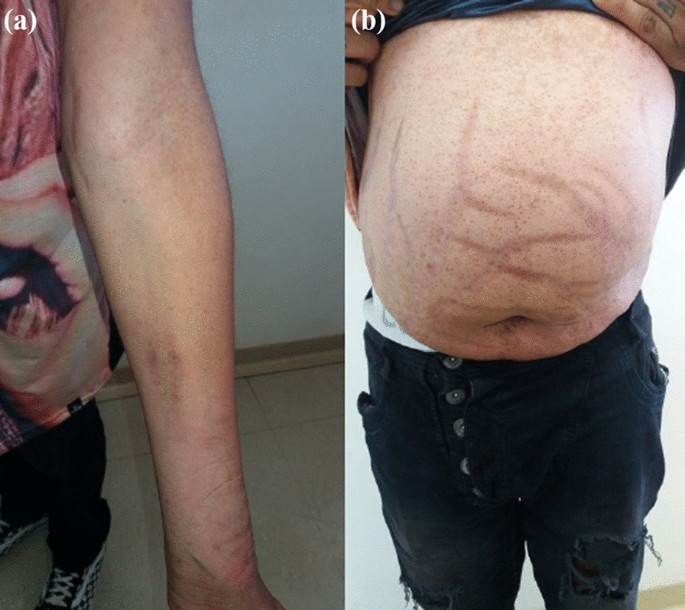
On physical examination, there were no lentiginous skin areas or blue nevi; however, wide violet streaks were observed on the upper limbs and abdomen, with plethora and increased fat in the temporal facial region and hump (Fig. 2a, b), limb ecchymosis, hypotrophy of the arms and thighs, central obesity and kyphoscoliosis. Systemic blood pressure (sitting) was 150 × 90 mmHg, BMI was 26.09 kg/m2, and waist circumference was 99 cm, with no reported reduction in height, maintained at 1.55 m.
Fig. 2
Changes in the physical examination. a Violet streaks on the upper limbs, b Violet streaks on abdomen
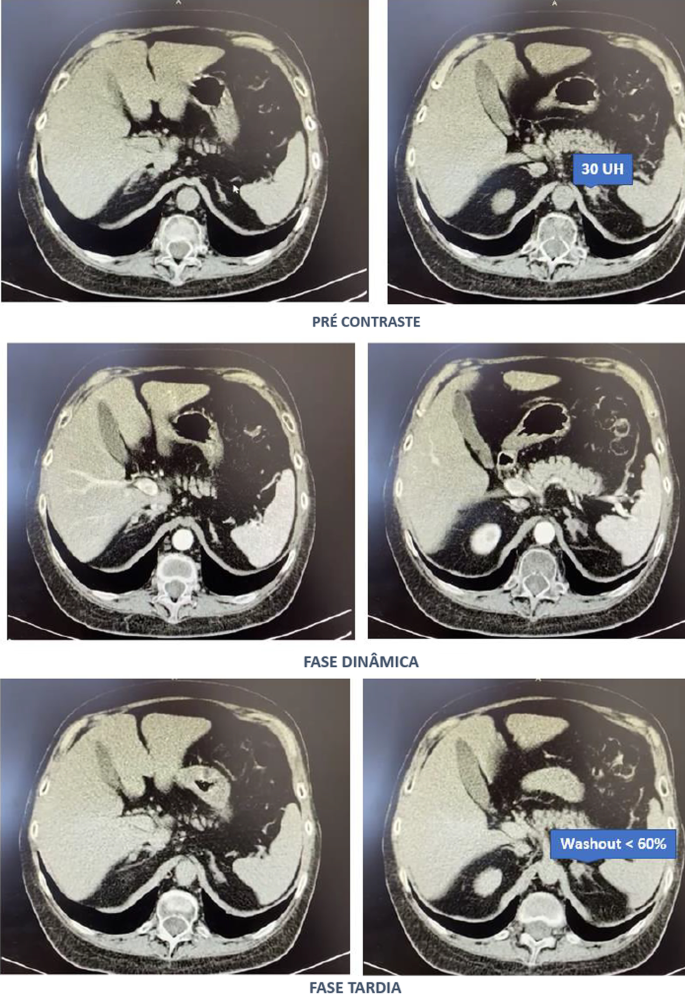
Computed tomography of the abdomen with adrenal protocol performed on 08/13/2020 characterized isodense nodular formation in the body of the left adrenal and in the lateral portion of the right adrenal, measuring 1.5 cm and 0.6 cm, respectively. The lesions had attenuation of approximately 30 HU, showing enhancement by intravenous contrast, with an indeterminate washout pattern in the late phase after contrast (< 60%) (Fig. 3).
After contact with the interventional radiology of the Hospital of Federal University of Juiz de Fora, catheterization of adrenal veins was performed on 10/2020; however, it was not possible to perform adequate lesion characterization due to obtaining serum cortisol levels that extrapolated the dilutional upper limit of the method (Table 3).
The calculation of the selectivity index was 6.63 (Reference Value (RV) > 3), confirming the good positioning of the catheter within the vessels during the procedure. The calculated lateralization index was 1.1296 (VR < 3), denoting bilateral hormone production. However, as aldosterone was not collected from a peripheral vein, it was not possible to obtain the contralateral rate and define whether there was contralateral suppression of aldosterone production [5].
Due to pending diagnoses for a better therapeutic decision and Cushing’s syndrome in clear evolution and causing organic damage, it was decided, after catheterization, to make changes in the patient’s drug prescription. Ketoconazole 400 mg per day was started, the dose of vitamin D was increased to 14,000 IU per week, and ramipril 5 mg per day was prescribed due to secondary hypertension. In addition, given the severity of osteoporosis, it was decided to replace previously prescribed alendronate with zoledronic acid.
Magnetic resonance imaging of the upper abdomen was performed on 06/19/2021, which demonstrated lobulated nodular thickening in the left adrenal gland with areas of decreased signal intensity in the T1 out-phase sequence, denoting the presence of fat, and homogeneous enhancement using contrast, measuring approximately 1.7 × 1.5 × 1.3 cm, suggestive of an adenoma. There was also a small nodular thickening in the lateral arm of the right adrenal, measuring approximately 0.8 × 0.6 cm, which was difficult to characterize due to its small dimensions and nonspecific appearance.
PPNAD or carcinoma became an important etiological hypothesis for the case described when comparing the epidemiology in a young man and the clinical-laboratory-imaging findings of the differential diagnoses. According to a dialog with the patient and family, the group of experts opted for unilateral glandular surgical resection on the left side (11/11/2021), where more significant changes were visualized, as there was a possibility of malignancy in a young patient and to avoid a definitive adrenal insufficiency condition because of bilateral adrenalectomy. This would first allow the analysis of the material and follow-up of the evolution of the condition with the permanence of the contralateral gland.
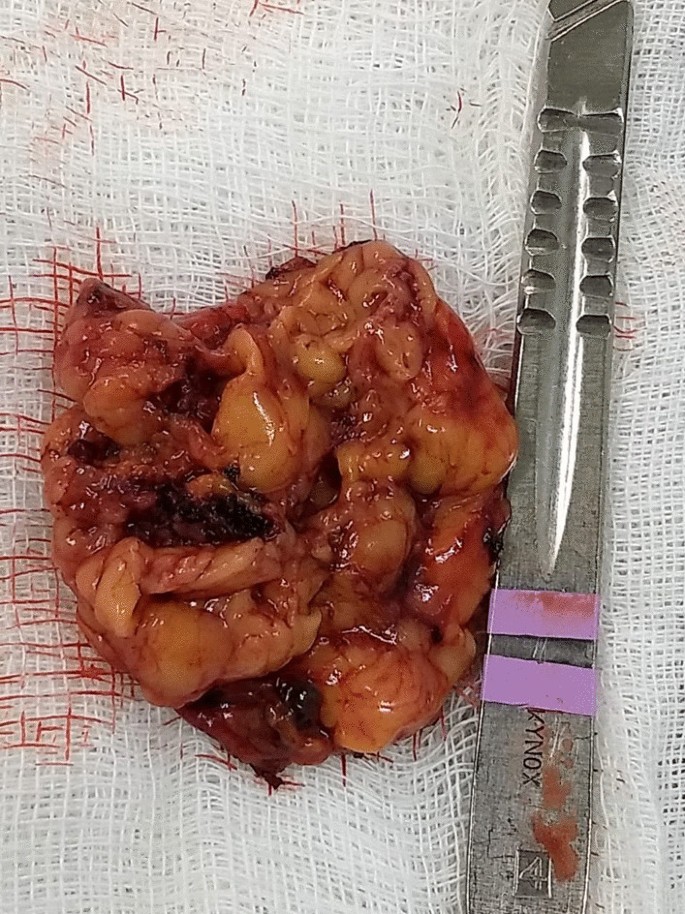
In the macroscopic analysis of the adrenalectomy specimen, adrenal tissue weighing 20 g and measuring 9.3 × 5.5 × 2.0 cm was described, completely surrounded by adipose tissue. The gland has a multinodular surface and varies between 0.2 and 1.6 cm in thickness, showing a cortex of 0.1 cm in thickness and a medulla of 1.5 cm in thickness (Fig. 4).
The microscopic analysis described the expansion of the zona fasciculate, with the formation of multiple nonencapsulated nodules composed of polygonal cells with ample and eosinophilic cytoplasm and frequent depletion of intracytoplasmic lipid content. No areas of necrosis or mitotic activity were observed. The histopathological picture is suggestive of cortical pigmented micronodular hyperplasia of the adrenal gland.
For the final etiological definition and an indication of contralateral adrenalectomy, which could be unnecessary and would avoid chronic corticosteroid therapy, or else, it would be necessary to protect the patient from future complications with the maintenance of the disease in the right adrenal gland, it would be essential to search for mutations in the PRKAR1A, PDE11A, PDE8B and PRKACA genes [15]; however, such genetic analysis is not yet widely available, and the impossibility of carrying it out at the local level did not allow a complete conclusion of the case.
Discussion
Through the clinical picture presented and the research of several secondary causes for osteoporosis, it was possible to arrive at the diagnosis of Cushing syndrome [6]. There was symptomatic independent ACTH hypercortisolism, manifested by typical phenotypic changes, severe secondary osteoporosis, and arterial hypertension in a young patient.
The diagnosis of Cushing’s syndrome is always challenging, given the presence of confounding factors such as the following:
Frequent and, even unknown, short- and long-term use of corticosteroids under different presentations;
Increase in the general population incidence of diabetes and obesity;
Screening tests with singularities for collection and individualized for different patient profiles.
It is important to note that the basal morning cortisol measurement is not the ideal test to assess hypercortisolism and is better applied to the assessment of adrenal insufficiency. However, the hypercortisolism of the case was unequivocal, and this test was also shown to be altered several times. As no test is 100% accurate, the current guidelines suggest the use of at least two first-line functional tests that focus on different aspects of the pathophysiology of the hypothalamic‒pituitary‒adrenal axis to confirm the hypercortisolism state: 24-hours cortisol, nocturnal salivary cortisol, morning serum cortisol after suppression with 1 mg of dexamethasone or after Liddle 1. Given that night-time salivary cortisol would require hospitalization, the other suggested tests were chosen, which are easier to perform in this context [7, 8].
Subsequently, tests were performed to determine the cause of hypercortisolism, such as serum ACTH levels and adrenal CT. The suppressed ACTH denoted the independence of its action. CT showed bilateral adrenal nodules with more severe features: solid lesion, attenuation > 10 UI on noncontrast images, and contrast washout speed < 60% in 10 minutes. In this case, it is essential to make a broad clinical decision and dialog with the patient to weigh and understand the risks and benefits of surgical treatment [9].
Among the main diagnostic hypotheses for the differential diagnosis of bilateral adrenal hyperplasia are primary bilateral macronodular adrenal hyperplasia, McCune–Albright syndrome (MAS) and bilateral primary pigmented nodular hyperplasia (PPNAD) isolated or associated with Carney’s complex. Another possibility would be bilateral adrenocorticotropic hormone (ACTH)-dependent macronodular hyperplasia secondary to long-term adrenal stimulation in patients with Cushing’s disease (ACTH-secreting pituitary tumor) or ectopic ACTH production, but the present case did not present with ACTH elevation.
Primary macronodular adrenal hyperplasia (nodules > 1 cm) predominates in women aged 50–60 years and may also be detected in early childhood (before 5 years) in the context of McCune–Albright syndrome. Most cases are considered sporadic; however, there are now several reports of familial cases whose presentation suggests autosomal dominant transmission. Several pathogenic molecular causes were identified in the table, indicating that it is a heterogeneous disease [10]. The pathophysiology occurs through the expression of anomalous ectopic hormone receptors or amplified eutopic receptors in the adrenals. It usually manifests in an insidious and subclinical way, with cortisol secretion mediated through receptors for gastric inhibitory peptide (GIP), vasopressin (ADH), catecholamines, interleukin 1 (IL-1), leptin, luteinizing hormone (LH), serotonin or others. Nodular development is not always synchronous or multiple; thus, hypercortisolism only manifests when there is a considerable increase in the number of adrenocortical cells, with severe steroidogenesis observed by cortisoluria greater than 3 times the upper limit of normal. Patients with mild Cushing’s syndrome should undergo screening protocols to identify aberrant receptors, as this may alter the therapeutic strategy. If there is evidence of abnormal receptors, treatment with beta-blockers is suggested for patients with beta-adrenergic receptors or with gonadotropin-releasing hormone (GnRH) agonists (and sex steroid replacement) for patients with LH/hCG receptors. In patients in whom aberrant hormone receptors are not present or for whom no specific pharmacological blockade is available or effective, the definitive treatment is bilateral adrenalectomy, which is known to make the patient dependent on chronic corticosteroid therapy [11]. Studies have shown the effectiveness of unilateral surgery in the medium and long term, opting for the resection of the adrenal gland of greater volume and nodularity by CT, regardless of the values obtained by catheterization of adrenal veins, but with the possibility of persistence or recurrence in the contralateral gland. Another possibility would be total unilateral adrenalectomy associated with subtotal contralateral adrenalectomy [12].
In McCune–Albright syndrome (MAS), there are activating mutations in the G-protein GNAS1 gene, generating autonomic hyperfunction of several tissues, endocrine or not, and there may be, for example, a constant stimulus similar to ACTH on the adrenal gland. In this case, pituitary levels of ACTH are suppressed, and adrenal adenomas with Cushing’s syndrome appear. Hypercortisolism may occur as an isolated manifestation of the syndrome or be associated with the triad composed of polyostotic fibrous dysplasia, café au lait spots with irregular borders and gonadal hyperfunction with peripheral precocious puberty. The natural history of Cushing’s syndrome in McCune-Albright syndrome (MAS) is heterogeneous, with some children evolving with spontaneous resolution of hypercortisolism, while others have a more severe condition, eventually requiring bilateral adrenalectomy [13].
PPNAD predominates in females, in people younger than 30 years, multiple and small (< 6 mm) bilateral pigmented nodules (surrounded by atrophied cortex), which can reach 1.5 cm in adulthood, with family genetic inheritance (66%) or sporadic inheritance (33%), and as part of the Carney complex reported in 40% of cases. In 70% of cases, inactivating mutations are identified in the PKA regulatory 1-alpha subunit (PRKAR1A), a tumor suppressor gene [14]. Osteoporosis is often associated with this condition [15]. One test that can distinguish patients with PPNAD from other primary adrenocortical lesions is cortisoluria after sequential suppression with low- and high-dose dexamethasone. In contrast to most patients with primary adrenocortical disease, who demonstrate no change in urinary cortisol, 70% of PPNAD patients have a paradoxical increase in urinary cortisol excretion [16]. The treatment of choice for PPNAD is bilateral adrenalectomy due to the high recurrence rate for primary adrenal disease [17].
Carney complex is a multiple neoplastic syndrome with autosomal dominant transmission, characterized by freckle-like cutaneous hyperpigmentation (lentiginosis), endocrine tumors [(PPNAD), testicular and/or thyroid tumors and acromegaly] and nonendocrine tumors, including cutaneous, cardiac, mammary, and osteochondral myxomas, among others. In the above case, the transthoracic echocardiogram of the patient on 03/18/2021 showed cavities of normal dimensions, preserved systolic and diastolic functions, no valve changes and no lentiginous skin areas and blue nevi, making the diagnosis of the syndrome less likely. The definitive diagnosis of Carney requires two or more main manifestations. Several related clinical components may suggest the diagnosis but not define it. The diagnosis can also be made if a key criterion is present and a first-degree relative has Carney or an inactivating mutation of the gene encoding PRKAR1A [18].
The adenoma is usually small in size (< 3 cm), similar to the nodules in this case; however, it is usually unilateral, with an insidious and mild evolution, especially in adult women over 35 years of age, producing only 1 steroid class. Carcinomas are usually large (> 6 cm), and only 10% are bilateral. They should be suspected mainly when the tumor presents with hypercortisolism associated with hyperandrogenism. They have a bimodal age distribution, with peaks in childhood and adolescence, as well as at the end of life [3].
Conclusion
Early identification of Cushing’s syndrome, with measures based on the assessment of risks and benefits, remains the best way to prevent progression and reduce morbidity [2]. After 6 months of drug inhibition of steroidogenesis, blood pressure control and anti-osteoporotic therapy, the objective was to minimize the levels and deleterious metabolic effects of hypercortisolism, which could also harm the surgical procedure in the short and long term through infections, dehiscence, nonimmediate bed mobilization and cardiovascular events. Unilateral adrenalectomy was chosen, given the possibility of malignancy in a young patient and to avoid definitive surgical adrenal insufficiency if the adrenalectomy was bilateral. Despite the unavailability of genetic analysis for a precise etiological definition, it is possible to take efficient measures to avoid unnecessary consequences or damage.
Pedro AO, Plapler PG, Szejnfeld VL. Manual brasileiro de osteoporose: orientações práticas para os profissionais de saúde. 1st ed. São Paulo: Editora Clannad; 2021. ISBN 978-65-89832-00-3.
Naguib R, Elkemary EZ, Elsharkawi KM. The severity of bone loss: a comparison between Cushing’s disease and Cushing’s syndrome. J Endocrinol Metab. 2023;13(1):33–8. https://doi.org/10.14740/jem857.
Wang D, Dang CX, Hao YX, Yu X, Liu PF, Li JS. Relationship between osteoporosis and Cushing syndrome based on bioinformatics. Medicine (Baltimore). 2022;101(43): e31283.
Williams TA, Reincke M. Management of Endocrine Disease: diagnosis and management of primary aldosteronism: the Endocrine Society guideline 2016 revisited. Eur J Endocrinol. 2018;179(1):R19–29. https://doi.org/10.1530/EJE-17-0990.
Compston J, Cooper A, Cooper C, Gittoes N, Gregson C, Harvey N, National Osteoporosis Guideline Group (NOGG), et al. UK clinical guideline for the prevention and treatment of osteoporosis. Arch Osteoporos. 2017;12(1):43. https://doi.org/10.1007/s11657-017-0324-5.
Hsiao HP, Kirschner LS, Bourdeau I, Keil MF, Boikos SA, Verma S, et al. Clinical and genetic heterogeneity, overlap with other tumor syndromes, and atypical glucocorticoid hormone secretion in adrenocorticotropin-independent macronodular adrenal hyperplasia compared with other adrenocortical tumors. J Clin Endocrinol Metab. 2009;94(8):2930–7. https://doi.org/10.1210/jc.2009-0516.
Stratakis CA, Sarlis N, Kirschner LS, Carney JA, Doppman JL, Nieman LK, et al. Paradoxical response to dexamethasone in the diagnosis of primary pigmented nodular adrenocortical disease. Ann Intern Med. 1999;131(8):585–91. https://doi.org/10.7326/0003-4819-131-8-199910190-00006.
Almeida MQ, Stratakis CA. Carney complex and other conditions associated with micronodular adrenal hyperplasias. Best Pract Res Clin Endocrinol Metab. 2010;24(6):907–14. https://doi.org/10.1016/j.beem.2010.10.006.
Serviço de Endocrinologia, Hospital Universitário da Universidade Federal de Juiz de Fora, Juiz de Fora, Minas Gerais, Brazil
Bárbara Oliveira Reis, Christianne Toledo Sousa Leal, Danielle Guedes Andrade Ezequiel, Ana Carmen dos Santos Ribeiro Simões Juliano, Flávia Lopes de Macedo Veloso, Leila Marcia da Silva, Lize Vargas Ferreira, Mariana Ferreira & Gabriel Zeferino De Oliveira Souza
Contributions
All the authors contributed to the conception and design of the work and have approved the submitted version. All authors read and approved the final manuscript.
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Surgical removal of adrenal gland.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Cushing’s syndrome (CS) shows diverse signs such as centripetal obesity, moon face, and buffalo hump, which can complicate the diagnosis. Facial features including eyelid edema, as an underrecognized sign, can be diagnostic clues for an excess of corticoids in a CS patient.
A 49-year-old woman presented with amenorrhea and weight gain that had continued for 2 years. Her medical history was dyslipidemia, hypertension, and osteoporosis. Physical examination revealed eyelid edemas (Figure 1A), moon face, buffalo hump, abdominal purple striae, and centripetal obesity (body mass index (BMI), 30.8 kg/m2). Basal plasma adrenocorticotropin was undetectable and serum cortisol level was high (16.9 μg/dl) without circadian rhythms. Free cortisol level in a 24-h urine collection was elevated (158.7 μg/day). Overnight administration of dexamethasone (1 mg) did not reduce serum cortisol level (17.4 μg/dl). Magnetic resonance imaging suggested bilateral adenomas. We made a diagnosis of adrenal Cushing’s syndrome (CS). Since 131l-adosterol scintigraphy showed specific uptake in the left adrenal gland, left adrenalectomy was laparoscopically performed. Histopathology of the tumor was compatible with adrenocortical adenoma. Three months after surgery, her BMI decreased to 25.0 kg/m2 and eyelid edemas were ameliorated (Figure 1B).
(A) Bilateral eyelid edemas due to Cushing’s syndrome are shown. (B) These findings were improved three months after surgery for left adrenal adenomas
Eyelid edema, in addition to centripetal obesity, moon face, and buffalo hump, is also a significant sign of CS; however, it has scarcely been reported in countries other than Japan.1, 2 Increased capillary permeability, insufficient venous return due to muscle atrophy, and sodium retention due to mineralocorticoid actions conceivably cause edema in CS.
AUTHORS’ CONTRIBUTIONS
KY wrote the first draft and managed all the submission processes. KO and KH contributed to the clinical management of the patient. FO organized the writing the manuscript.
ACKNOWLEDGMENT
None.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ETHICAL APPROVAL
Written informed consent was obtained from the patient to publish this case report.
1Lacroix A, Feelders RA, Stratakis CA, Nieman LK. Cushing’s syndrome. Lancet. 2015; 386: 913- 927.
2Komiya I, Takasu N, Ohara N, et al. Forty-one cases of Cushing’s syndrome: a comparison between Cushing’s syndrome (adrenal adenoma) and Cushing’s disease (adrenal hyperplasia). Nihon Naibunpi Gakkai Zasshi. 1992; 68: 607- 622.
Cases of adrenocorticotropic hormone (ACTH)-independent Cushing’s syndrome are often caused by unilateral tumors in the adrenal glands, but Indian researchers have now reported a rare case where the condition was caused by tumors in both adrenal glands.
Fewer than 40 cases of bilateral tumors have been reported so far, but an accurate diagnosis is critical for adequate and prompt treatment. Sampling the veins draining the adrenal glands may be a good way to diagnose the condition, researchers said.
Cushing’s syndrome, a condition characterized by excess cortisol in circulation, can be divided into two main forms, depending on ACTH status. Some patients have tumors that increase the amount of ACTH in the body, and this hormone will act on the adrenal glands to produce cortisol in excess. Others have tumors in the adrenal glands, which produce excess cortisol by themselves, without requiring ACTH activation. This is known as ACTH-independent Cushing’s syndrome.
Among the latter, the disease is mostly caused by unilateral tumors — in one adrenal gland only — with cases of bilateral tumors being extremely rare in this population.
Now, researchers reported the case of a 31-year-old Indian woman who developed ACTH-independent Cushing’s syndrome because of tumors in both adrenal glands.
The patient complained of weight gain, red face, moon face, bruising, and menstrual irregularity for the past two years. She recently had been diagnosed with high blood pressure and had started treatment the month prior to the presentation.
A physical examination confirmed obesity in her torso, moon face, buffalo hump, thin skin, excessive hair growth, acne, swollen legs and feet, and skin striae on her abdomen, arms, and legs.
Laboratory examinations showed that the woman had an impaired tolerance to glucose, excess insulin, and elevated cortisol in both the blood and urine. Consistent with features of Cushing’s syndrome, cortisol levels had no circadian rhythm and were non-responsive to a dexamethasone test, which in normal circumstances lowers cortisol production.
Because ACTH levels were within normal levels, researchers suspected an adrenal tumor, which led them to conduct imaging scans.
An abdominal computed tomography (CT) scan showed adrenal adenomas in both adrenal glands (right: 3.1 cm × 2.0 cm × 1.9 cm; left: 2.2 cm × 1.9 cm × 2.1 cm). A magnetic resonance imaging (MRI) scan showed that the pituitary gland (which normally produces ACTH) was normal.
To determine whether both adrenal tumors were producing cortisol, researchers sampled the adrenal veins and compared their cortisol levels to those of peripheral veins. They found that the left adrenal gland was producing higher amounts of cortisol, thought the right adrenal gland was also producing cortisol in excess.
“Our case indicates that adrenal vein [blood] sampling might be useful for obtaining differential diagnoses” in cases of Cushing’s syndrome, researchers stated. Also, they may help design a surgical plan that makes much more sense.”
The tumors were surgically removed — first the left, and three months later the right — which alleviated many of her symptoms. She also started prednisolone treatment, which helped resolve many disease symptoms.
“Bilateral cortisol-secreting tumors are a rare cause of Cushing’s syndrome,” researchers said. So when patients present bilateral adrenal lesions, “it is crucial to make a definitive diagnosis before operation since various treatments are prescribed for different causes,” they said.
The team recommends that in such cases the two tumors should not be removed at the same time, as this approach may cause adrenal insufficiency and the need for glucocorticoid replacement therapy.



-bilateral-multilobar-pneumonia")
-depicting-an-8x8-cm-hematoma-(arrow)-involving-the-left-psoas-and-iliacus-muscles")
-of-the-brain-depicting-a-1.3x1.3x3.2-cm-pituitary-macroadenoma-(star)")