A new test developed by University of Manchester and NHS scientists could revolutionise the way children with growth hormone deficiency are diagnosed.
Children suspected of having GHD – which cause growth to slow down or stop and other serious physical problems—currently require a test involving fasting for up to 12 hours.
The fasting is followed by an intravenous infusion in hospital and up to 10 blood tests over half a day to measure growth hormone production.
Because the current test is unreliable, it often has to be done twice before growth hormone injections can be prescribed.
Now the discovery—which the team think could be available within 2 to 5 years -could reduce the process to a single blood test, freeing up valuable time and space for the NHS.
Dr. Adam Stevens from The University of Manchester and Dr. Philip Murray from Manchester University NHS Foundation Trust, were part of the team whose results are published in JCI Insight today.
Dr. Stevens said: “We think this is an important development in the way doctors will be able to diagnose growth hormone deficiency – a condition which causes distress to many thousands of children in the UK
“This sort of diagnostic would not be available even a few years ago but thanks to the enormous computing power we have, and advances in genetics, it is now possible for this aspect of care to be made so much easier for patients – and the NHS.
“These volume of data involved is so huge and complicated that traditional data-processing application software is inadequate to deal with it.”
Comparing data from 72 patients with GHD and 26 healthy children, they used high powered computers to examine 30,000 genes—the full gene expression- of each child.
A sophisticated mathematical technique called Random Forest Analysis analysed around three million separate data points to compare different gene patterns between the children with and without GHD.
The research identified 347 genes which when analysed with the computer algorithm can determine whether a child has GHD or not and thus whether they will benefit from treatment.
Growth hormone deficiency (GHD) occurs when the pituitary gland—which is size of a pea- fails to produce enough growth hormone. It more commonly affects children than adults.
Many teenagers with GHD have poor bone strength, fatigue and lack stamina as well as depression, lack of concentration, poor memory and anxiety problems.
GHD occurs in roughly 1 in 5,000 people. Since the mid-1980s, synthetic growth hormones have been successfully used to treat children—and adults—with the deficiency.
Dr. Murray added: “This study provides strong proof of concept, but before it is in a position to be adopted by the NHS, we must carry out a further validation exercise which will involve comparing our new diagnostic with the existing test.
“Once we have crossed that hurdle, we hope to be in a position for this to be adopted within 2 to 5 years – and that can’t come soon enough for these children.”
Child Growth Foundation manager Jenny Child’s daughter has Growth Hormone Deficiency.
She said: Growth Hormone Deficiency isn’t just about growth, as lack of growth hormone impacts the child in many ways, such as lack of strength and they can find it difficult to keep up physically with their peers. It impacts the child’s self-esteem as they are often treated as being much younger, because of their size. Growth hormone treatment allows the child to grow to their genetic potential.
“A growth hormone stimulation test can be very daunting for both child and parents. The test can make the child feel quite unwell and they can experience headaches, nausea and unconsciousness through hypoglycaemia.”
More information: Philip G. Murray et al. Transcriptomics and machine learning predict diagnosis and severity of growth hormone deficiency, JCI Insight (2018). DOI: 10.1172/jci.insight.93247
Thymic neuroendocrine tumor as a cause of Cushing syndrome is extremely rare in children.
Case presentation
We report a case of a 10-year-old girl who presented with typical symptoms and signs of hypercortisolemia, including bone fractures, growth retardation, and kidney stones. The patient was managed with oral ketoconazole, during which she experienced adrenal insufficiency, possibly due to either cyclic adrenocorticotropic hormone (ACTH) secretion or concurrent COVID-19 infection. The patient underwent a diagnostic work-up which indicated the possibility of an ACTH-secreting pituitary neuroendocrine tumor. However, after a transsphenoidal surgery, the diagnosis was not confirmed on histopathological examination. Subsequent bilateral inferior petrosal sinus sampling showed strong indications of the presence of ectopic ACTH syndrome. Detailed rereading of functional imaging studies, including 18F-FDG PET/MRI and 68Ga DOTATOC PET/CT, ultimately identified a small lesion in the thymus. The patient underwent videothoracoscopic thymectomy that confirmed a neuroendocrine tumor with ACTH positivity on histopathological examination.
Conclusion
This case presents some unique challenges related to the diagnosis, management, and treatment of thymic neuroendocrine tumor in a child. We can conclude that ketoconazole treatment was effective in managing hypercortisolemia in our patient. Further, a combination of functional imaging studies can be a useful tool in locating the source of ectopic ACTH secretion. Lastly, in cases of discrepancy in the results of stimulation tests, bilateral inferior petrosal sinus sampling is highly recommended to differentiate between Cushing disease and ectopic ACTH syndrome.
In children above seven years of age, the majority of pediatric Cushing syndrome (CS) cases are caused by a pituitary neuroendocrine tumors (PitNET). However, a differential diagnosis of hypercortisolemia in children is often challenging concerning the interpretation of stimulation tests and the fact that up to 50% of PitNET may not be detected on magnetic resonance imaging (MRI) [1]. An ectopic adrenocorticotropic hormone (ACTH) syndrome (EAS) is extremely rare in children. Its diagnosis is often missed or confused with Cushing disease (CD) [2]. Most ACTH-secreting tumors originate from bronchial or thymic neuroendocrine tumors (NETs), or less commonly, from NETs in other locations. To diagnose EAS, specific functional imaging studies are often indicated to elucidate the source of ACTH production.
Pharmacotherapy may be used before surgery to control hypercortisolemia and its symptoms/signs, or in patients in whom the source of hypercortisolism has not been found (e.g., EAS), or surgery failed. Ketoconazole or metyrapone, as adrenal steroidogenesis blockers, were found to be very efficient, although they exhibit side effects [3].
Furthermore, cyclic secretion of ACTH followed by fluctuating plasma cortisol levels is extremely rare in children, including those with EAS [4, 5]. Therefore, in cyclic EAS, the use of steroid inhibitors or acute illness or trauma can be associated with adrenal insufficiency, which can be life-threatening. Here we describe the clinical features, laboratory and radiological investigations, results, management, and clinical outcome of a 10-year-old girl with a thymic NET presenting with ACTH secretion.
Case presentation
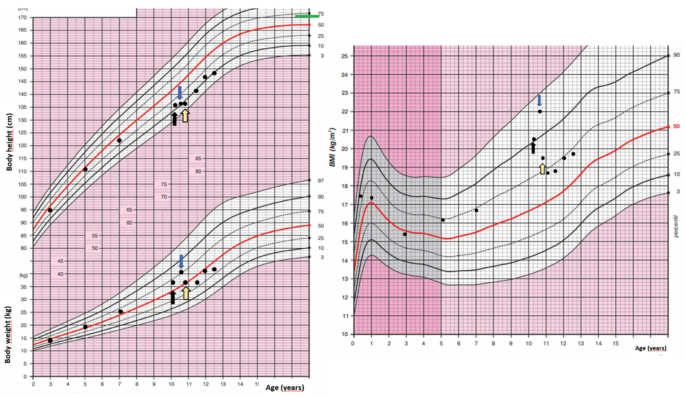
A 10-year-old girl was acutely admitted to our university hospital for evaluation of facial edema and macroscopic hematuria in May 2021. A day before admission, she presented to the emergency room for dysuria, pollakiuria, nausea, and pain in her right lower back. Over the past year she had experienced excessive weight gain with increased appetite and growth retardation (Fig. 1). Her height over three years had shifted from the 34th to the 13th centile (Fig. 1). Her parents noticed facial changes, pubic hair development, increased irritability, and moodiness.
Fig. 1
Body weight, body height, and body mass index development of the case patient. The black arrow indicates the first presentation, the blue arrow indicates the start of ketoconazole treatment and the yellow arrow indicates the time of thymectomy. Mid-parental height is indicated by the green line
At admission, she was found to have a moon face with a plethora, few acne spots on forehead, as well as facial puffiness. In contrast to slim extremities, an abnormal fat accumulation was observed in the abdomen. Purple striae were present on abdomen and thighs. She did not present with any bruising, proximal myopathy, or edema. On physical examination, she was prepubertal, height was 135 cm (13th centile), and weight was 37 kg (69th centile) with a BMI of 20.4 kg/m2 (90th centile). She developed persistent hypertension. Her past medical history was uneventful except for two fractures of her upper left extremity after minimal trips one and three years ago, both treated with a caste. Apart from hypothyroidism on the maternal side, there was no history of endocrine abnormalities or tumors in the family.
In the emergency room, the patient was started on sulfonamide, pain medication, and intravenous (IV) fluids. Her hypertensive crises were treated orally with angiotensin-converting enzyme inhibitor or with a combination of adrenergic antagonists and serotonin agonists administered IV. Hypokalemia had initially been treated with IV infusion and then with oral potassium supplements. A low serum phosphate concentration required IV management. The initial investigation carried out in the emergency room found hematuria with trace proteinuria. Kidney ultrasound showed a 5 mm stone in her right ureter with a 20 mm hydronephrosis. She did not pass any kidney stones, however, fine white sand urine analysis reported 100% brushite stone.
Hypercortisolemia was confirmed by repeatedly increased 24-hour urinary free cortisol (UFC), (5011.9 nmol/day, normal range 79.0-590.0 nmol/day). Her midnight cortisol levels were elevated (961 nmol/l, normal range 68.2–537 nmol/l). There was no suppression of serum cortisol after 1 mg overnight dexamethasone suppression test (DST) or after low-dose DST (LDDST). An increased morning plasma ACTH (30.9 pmol/l, normal range 1.6–13.9 pmol/) suggested ACTH-dependent hypercortisolemia. There was no evidence of a PitNET on a 1T contrast-enhanced MRI. The high-dose DST (HDDST) did not induce cortisol suppression (cortisol 1112 nmol/l at 23:00, cortisol 1338 nmol/l at 8:00). Apart from the kidney stone, a contrast-enhanced computed tomography (CT) of her neck, chest, and abdomen/pelvis did not detect any lesion. Various tumor markers were negative and the concentration of chromogranin A was also normal.
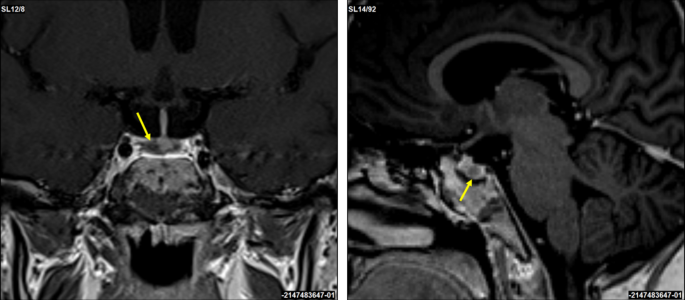
A corticotropin-releasing hormone (CRH) stimulation test induced an increase in serum cortisol by 32% at 30 min and ACTH concentration by 67% at 15 min (Table 1). A 3T contrast-enhanced MRI scan of the brain identified a 3 × 2 mm lesion in the lateral right side of the pituitary gland (Fig. 2). An investigation of other pituitary hormones was unremarkable. Apart from low serum potassium (minimal level of 2.8 mmol/l; normal range 3.3–4.7 mmol/l) and phosphate (0.94 mmol/l; normal range 1.28–1.82 mmol/l) concentrations, electrolytes were normal. The bone mineral density assessed by whole dual-energy X-ray absorptiometry was normal.
Fig. 2
Coronal and sagittal 3T contrast-enhanced brain MRI scans. A suspected 3 × 2 mm lesion in the lateral right side of the pituitary gland (yellow arrows)
The patient was presented at the multidisciplinary tumor board and it was decided that she undergoes transsphenoidal surgery for the pituitary lesion. No PitNET was detected on histopathological examination and no favorable biochemical changes were noted after surgery. After the patient recovered from surgery, subsequent bilateral inferior petrosal sinus sampling (BIPSS) confirmed EAS as the maximum ratio of central to peripheral ACTH concentrations was only 1.7. During the investigation for tumor localization, she was started on ketoconazole treatment (300 mg/day) to alleviate symptoms and signs of hypercortisolism. Treatment with ketoconazole had a beneficial effect on patient health (Fig. 1). There was a weight loss of 2 kg in a month, a disappearance of facial plethora, and a decrease in vigorous appetite. Her liver function tests remained within the normal range.
Table 1 Result of corticotropin-releasing hormone stimulation test
The 24-hour UFC excretion normalized three weeks after ketoconazole initiation. However, six weeks after continuing ketoconazole therapy (400 mg/day), the patient complained of nausea, vomiting, and diarrhea. She was found to have adrenal insufficiency with a low morning serum cortisol of 10.70 nmol/l (normal range 68.2–537 nmol/l) and salivary cortisol concentrations < 1.5 nmol/l (normal range 1.7–29 nmol/l). She was also found to be positive for COVID-19 infection. Ketoconazole treatment was stopped and our patient was educated to take stress steroids in case of persisting or worsening symptoms. Her clinical status gradually improved and steroids were not required.
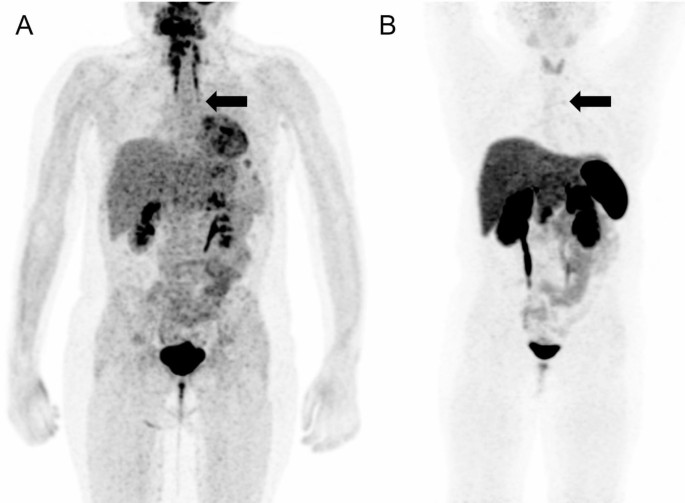
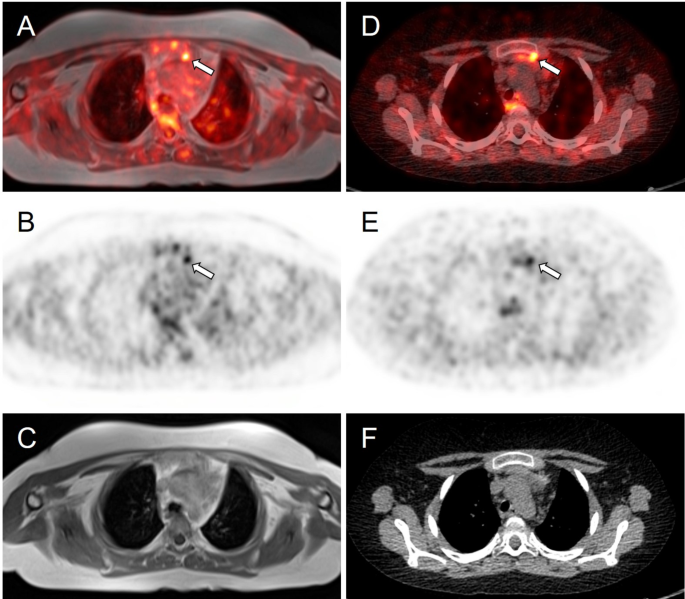
Meanwhile, whole-body fluorine-18 fluorodeoxyglucose positron emission tomography (18F-FDG PET)/MRI was performed with no obvious hypermetabolic lesion suspicious of a tumor. No obvious accumulation was detected on 68Ga-DOTATOC PET/CT images (Fig. 3). However, a subsequent careful and detailed re-review of the images detected a discrete lesion on 18F-FDG PET/MRI and 68Ga-DOTATOC PET/CT scans in the left anterior mediastinum, in the thymus (Fig. 4).
Fig. 3
18F-FDG PET/MRI (A) and 68Ga-DOTATOC (B) PET/CT scans. Whole body MIP reconstructions. Subtle correspondent focal hyperactivity in the left mediastinum (black arrow). The 18F-FDG PET/MRI image courtesy of Prof. Jiri Ferda, MD, PhD, Clinic of the Imaging Methods, University Hospital Plzen, Czech Republic
Axial slices of PET/MRI (A–C) and 68Ga-DOTATOC (D–F) PET/CT scans. Subtle correspondent focal hyperactivity in the left mediastinum (white arrow). No obvious finding on MRI (C) and CT (F) scans. The FDG PET/MRI image courtesy of Prof. Jiri Ferda, MD, PhD, Clinic of the Imaging Methods, University Hospital Plzen, Czech Republic
Three weeks after the episode of adrenal insufficiency and being off ketoconazole treatment, our patient´s pre-surgery laboratory tests showed slightly low morning cortisol 132 nmol/l with surprisingly normal ACTH 2.96 pmol/l (normal range 1.6–13.9 pmol/). Given the upcoming surgery, she was initiated on a maintenance dose of hydrocortisone (15 mg daily = 12.5 mg/m2/day). Further improvement of cushingoid characteristics (improvement of facial plethora and moon face, weight loss) was noticed. Our patient underwent videothoracoscopic surgery, and a hyperplastic thymus of 80 × 70 × 15 mm with a 4 mm nodule was successfully removed. Tumor immunohistochemistry was positive for ACTH, chromogranin A, CD56, and synaptophysin. Histopathological findings were consistent with a well-differentiated NET grade 1. A subsequent genetic screening did not detect any pathogenic variant in the MEN1 gene.
After surgery, hydrocortisone was switched to a stress dose and gradually decreased to a maintenance dose. Antihypertensive medication was stopped and further weight loss was observed after thymectomy. Within a few weeks after the thoracic surgery, the patient entered puberty, her mood improved significantly, and potassium supplements were stopped. Finally, hydrocortisone treatment was stopped ten months after thymectomy.
Discussion and conclusions
The case presented here demonstrates a particularly challenging work-up of the pediatric patient with the diagnosis of CS caused by EAS due to thymic NET. Differentiating CD and EAS can sometimes be difficult, including the use of various laboratory and stimulation tests and their interpretation, as well as proper, often challenging, reading of functional imaging modalities, especially if a discrete lesion is present at an unusual location [1]. When using established criteria for Cushing disease (for the CRH test an increase of cortisol and/or ACTH by ≥ 20% or ≥ 35%, respectively, and a ≥ 50% suppression of cortisol for the HDDST) our patient presented discordant results. The CRH stimulation test induced an increase in cortisol by 32% and ACTH by 67% and the 3T MRI pointed to the right-side pituitary lesion, both to yield false positive results. The HDDST, on the other hand, did not induce cortisol suppression and was against characteristic findings for CD. We did not proceed with desmopressin testing, which also induces an excess ACTH and cortisol response in CD patients and has rarely been used in pediatric patients, except in those with extremely difficult venous access [6]. Recently published articles investigated the reliability of CRH stimulation tests and HDDST and both concluded that the CRH test has greater specificity than HDDST [7, 8]. Elenius et al. suggested optimal response criteria as a ≥ 40% increase of ACTH and/or cortisol (cortisol as the most specific measure of CD) during the CRH test and a ≥ 69% suppression of serum cortisol during HDDST [7]. Using these criteria, the CD would be excluded in our patient. To demonstrate that the proposed thresholds for the test interpretation widely differ, Detomas et al. proposed a ≥ 12% cortisol increase and ≥ 31% ACTH increase during the CRH test to confirm CD [8].
The fact that up to 50% of PitNET may not be detected on MRI [1] and that more than 20% of patients with EAS are reported to have pituitary incidentalomas [9] makes MRI somewhat unreliable in differentiating CD and EAS. However, finally, well-established and generally reliable BIPSS in our patient supported the diagnosis of EAS. Thus, BIPSS is considered a gold standard to differentiate between CD and EAS; however, it can still provide false negative results in cyclic CS if performed in the trough phase [10] or in vascular anomalies or false positive results as in a recent case of orbital EAS [11].
In children, the presence of thymus tissue may be misinterpreted as normal. Among other reports of thymic NET [12], Hanson et al. reported a case of a prepubertal boy in whom a small thymic NET was initially treated as normal thymus tissue on CT [13]. In our case, initially, the lesion was not detected on the 18F-FDG and 68Ga-DOTATOC PET scans. A small thymic NET was visible only after a detailed and careful re-reading of both PET scans. Although somatostatin receptor (SSR) PET imaging may be helpful in identifying ectopic CRH- or ACTH-producing tumors, there are still some limitations [13]. For example, in the study by Wannachalee et al., 68Ga-DOTATATE identified suspected primary lesions causing ECS in 65% of patients with previously occult tumors and was therefore concluded as a sensitive method for primary as well as metastatic tumors [14]. In our patient, the final correct diagnosis was based on the results of both PET scans. This is in full support of the article published by Liu et al. who concluded that 18F-FDG and SSR PET scans are complementary in determining the proper localization of ectopic ACTH production [15]. Additionally, it is worth noting that not all NETs stain positively for ACTH which may present a burden in its identification.
To control hypercortisolemia, both ketoconazole and metyrapone were considered in our patient. Due to the side effects of metyrapone on blood pressure, ketoconazole was started as a preferred option in our pediatric patient. A retrospective multicenter study concluded that ketoconazole treatment is effective with acceptable side effects, with no fatal hepatitis and adrenal insufficiency in 5.4% of patients [3]. During ketoconazole treatment, our patient developed adrenal insufficiency; however, it is impossible to conclude whether this was solely due to ketoconazole treatment or whether an ongoing COVID-19 infection contributed to the adrenal insufficiency or whether this was caused by a phase of lower or no ACTH secretion from the tumor often seen in patients with cyclic ACTH secretion. The patient’s cyclic ACTH secretion is highly probable since her morning cortisol was slightly lower and ACTH was normal, even after being off ketoconazole treatment for 3 weeks.
When retrospectively and carefully reviewing all approaches to the diagnostic and management care of our pediatric patient, it would be essential to proceed to BIPSS before any pituitary surgery, especially when obtaining discrepant results from stimulation tests, as well as detecting a discrete pituitary lesion (≤ 6 mm) as recommended by the current guidelines [16]. This was our first experience using ketoconazole in a young child, and although this treatment was associated with very good outcomes in treating hypercortisolemia, close monitoring, and family education on signs and symptoms of adrenal insufficiency are essential to recognizing adrenal insufficiency promptly in any patient with EAS, especially those presenting also with some other comorbidities or stress, here COVID-19 infection.
In conclusion, the pediatric patient here presenting with EAS caused by thymic NET needs very careful assessment including whether cyclic CS is present, the outline of a good management plan to use all tests appropriately and in the correct sequence, monitoring carefully for any signs or symptoms of adrenal insufficiency, and apply appropriate imaging studies, with experienced radiologists providing accurate readings. Furthermore, ketoconazole treatment was found to be effective in reducing the symptoms and signs of CS in this pediatric patient. Finally, due to the rarity of this disease and the challenging work-up, we suggest that a multidisciplinary team of experienced physicians in CS management is highly recommended.
Data availability
No datasets were generated or analysed during the current study.
Streuli R. A rare case of an ACTH/CRH co-secreting midgut neuroendocrine tumor mimicking Cushing’s disease. Endocrinol Diabetes Metab Case Rep. 2017;2017:17–58. ,Krull I, Brändle M, et al.
Karageorgiadis AS, Papadakis GZ, Biro J, et al. Ectopic adrenocorticotropic hormone and corticotropin-releasing hormone co-secreting tumors in children and adolescents causing cushing syndrome: a diagnostic dilemma and how to solve it. J Clin Endocrinol Metab. 2015;100(1):141–8.
Mi Q, Yin M-Z, Gao Y-J et al. Thymic atypical carcinoid with cyclical Cushing’s syndrome in a 7-year-old boy: a case report and review of the literature. Intern Med. 2014;4(5).
Moszczyńska E, Pasternak-Pietrzak K, Prokop-Piotrkowska M, et al. Ectopic ACTH production by thymic and appendiceal neuroendocrine tumors – two case reports. J Pediatr Endocrinol Metab. 2020;34(1):141–6.
Crock PA, Ludecke DK, Knappe UJ, et al. A personal series of 100 children operated for Cushing’s disease (CD): optimizing minimally invasive diagnosis and transnasal surgery to achieve nearly 100% remission including reoperations. J Pediatr Endocrinol Metab. 2018;31(9):1023–31.
Detomas M, Ritzel K, Nasi-Kordhishti I, et al. Outcome of CRH stimulation test and overnight 8 mg dexamethasone suppression test in 469 patients with ACTH-dependent Cushing’s syndrome. Front Endocrinol (Lausanne). 2022;13:955945.
Yogi-Morren D, Habra MA, Faiman C, et al. Pituitary MRI findings in patients with pituitary and ectopic ACTH-dependent Cushing syndrome: does a 6-mm pituitary tumor size cut-off value exclude ectopic ACTH syndrome? Endocr Pract. 2015;21(10):1098–103.
Albani A, Berr CM, Beuschlein F, et al. A pitfall of bilateral inferior petrosal sinus sampling in cyclic Cushing’s syndrome. BMC Endocr Disord. 2019;19(1):105.
Tan H, Chen D, Yu Y, et al. Unusual ectopic ACTH syndrome in a patient with orbital neuroendocrine tumor, resulted false-positive outcome of BIPSS: a case report. BMC Endocr Disord. 2020;20(1):116.
Ahmed MF, Ahmed S, Abdussalam A, et al. A rare case of ectopic adrenocorticotropic hormone syndrome (EAS) in an adolescent girl with a thymic neuroendocrine tumour. Cureus. 2024;16(8):e66615.
Hanson JA, Sohaib SA, Newell-Price J, et al. Computed tomography appearance of the thymus and anterior mediastinum in active Cushing’s syndrome. J Clin Endocrinol Metab. 1999;84:602–5.
Wannachalee T, Turcu AF, Bancos I, et al. The clinical impact of [68 Ga]-DOTATATE PET/CT for the diagnosis and management of ectopic adrenocorticotropic hormone – secreting Tumours. Clin Endocrinol (Oxf). 2019;91(2):288–94.
Liu Q, Zang J, Yang Y, et al. Head-to-head comparison of 68Ga-DOTATATE PET/CT and 18F-FDG PET/CT in localizing tumors with ectopic adrenocorticotropic hormone secretion: a prospective study. Eur J Nucl Med Mol Imaging. 2021;48(13):4386–95.
Flesiriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing’s disease: a guideline update. Lancet Diabetes Endocrinol. 2021;9(12):847–75.
The authors thank all the colleagues from the Thomayer University Hospital and Military University Hospital who were involved in the inpatient care of this patient.
Funding
This work was supported by the Charles University research program Cooperatio Pediatrics, Charles University, Third Faculty of Medicine, Prague.
Author information
Authors and Affiliations
Department of Children and Adolescents, Third Faculty of Medicine, Charles University, University Hospital Kralovske Vinohrady, Šrobárova 50, Prague, 100 34, Czech Republic
Irena Aldhoon-Hainerová
Department of Pediatrics, Thomayer University Hospital, Prague, Czech Republic
Irena Aldhoon-Hainerová
Department of Medicine, Military University Hospital, Prague, Czech Republic
Mikuláš Kosák
Third Department of Medicine, First Faculty of Medicine, Charles University, Prague, Czech Republic
Michal Kršek
Institute of Nuclear Medicine, First Faculty of Medicine, Charles University, General University Hospital, Prague, Czech Republic
David Zogala
Developmental Endocrinology, Metabolism, Genetics and Endocrine Oncology Affinity Group, Eunice Kennedy Shriver NICHD, NIH, Bethesda, MD, USA
Karel Pacak
Contributions
All authors made individual contributions to the authorship. IAH, MK, MK, and DZ were involved in the diagnosis and management of this patient. DZ was responsible for the patient´s imaging studies. IAH wrote the first draft of the manuscript. KP revised the manuscript critically. All authors reviewed and approved the final draft.
Signed informed consent was obtained from the patient and the patient´s parents for the publication of this case report and accompanying images.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
Paediatric endogenous Cushing syndrome is a rare condition with variable signs and symptoms of presentation. We studied a large cohort of paediatric patients with endogenous Cushing syndrome with the aim of describing anthropometric, clinical, and biochemical characteristics as well as associated complications and outcomes to aid diagnosis, treatment, and management.
Methods
In this prospective, multisite cohort study, we studied children and adolescents (≤18 years at time of presentation) with a diagnosis of Cushing syndrome. Patients had either received their initial diagnosis and evaluation at the Eunice Kennedy Shriver National Institute of Child Health and Human Development (Bethesda, MD, USA) or been referred from other centres in the USA or outside the USA. We collected participants’ clinical, biochemical, and imaging findings and recorded their post-operative course until their latest appointment.
Findings
Of 342 paediatric patients with a diagnosis of Cushing syndrome, 193 (56%) were female and 149 (44%) male. 261 (76%) patients had corticotroph pituitary neuroendocrine tumours (Cushing disease), 74 (22%) had adrenal-associated Cushing syndrome, and seven (2%) had ectopic Cushing syndrome. Patients were diagnosed at a median of 2 years (IQR 1·0–3·0) after the first concerning sign or symptom, and patients with adrenal-associated Cushing syndrome were the youngest at diagnosis (median 10·4 years [IQR 7·4–13·6] vs 13·0 years [10·5–15·3] for Cushing disease vs 13·4 years [11·0–13·7] for ectopic Cushing syndrome; p<0·0001). Body-mass index z-scores did not differ between the diagnostic groups (1·90 [1·19–2·34] for adrenal-associated Cushing syndrome vs 2·18 [1·60–2·56] for Cushing disease vs 2·22 [1·42–2·35] for ectopic Cushing syndrome; p=0·26). Baseline biochemical screening for cortisol and adrenocorticotropin at diagnosis showed overlapping results between subtypes, and especially between Cushing disease and ectopic Cushing syndrome. However, patients with ectopic Cushing syndrome had higher urinary free cortisol (fold change in median cortisol concentration from upper limit of normal: 15·5 [IQR 12·7–18·0]) than patients with adrenal-associated Cushing syndrome (1·5 [0·6–5·7]) or Cushing disease (3·9 [2·3–6·9]; p<0·0001). Common complications of endogenous Cushing syndrome were hypertension (147 [52%] of 281 patients), hyperglycaemia (78 [30%] of 260 patients), elevated alanine transaminase (145 [64%] of 227 patients), and dyslipidaemia (105 [48%] of 219 patients). Long-term recurrence was noted in at least 16 (8%) of 195 patients with Cushing disease.
Interpretation
This extensive description of a unique cohort of paediatric patients with Cushing syndrome has the potential to inform diagnostic workup, preventative actions, and follow-up of children with this rare endocrine condition.
Funding
Intramural Research Program, Eunice Kennedy Shriver National Institute of Child Health & Human Development, National Institutes of Health.
Introduction
Paediatric endogenous Cushing syndrome is a rare disorder accounting for 5–7% of all reported cases of endogenous Cushing syndrome.1, 2, 3 In children older than 5–7 years and adolescents, endogenous Cushing syndrome is most commonly caused by corticotroph pituitary neuroendocrine tumours (PitNETs) and is termed Cushing disease. By contrast, Cushing syndrome in children younger than 5 years is often associated with adrenal disorders and is termed adrenal-associated Cushing syndrome.4 Albeit rare, a third type termed ectopic Cushing syndrome is caused by neuroendocrine tumours outside the hypothalamic–pituitary axis that secrete adrenocorticotropin or corticotropin-releasing hormone.5, 6 Thus endogenous Cushing syndrome is caused by either adrenocorticotropin-dependent sources (pituitary or ectopic) or adrenocorticotropin-independent (adrenal) hypercortisolemia.
Patients with adults-onset Cushing syndrome typically present with weight gain, skin manifestations (striae, hirsutism, acne, and easy bruising), and abnormal fat deposition.7, 8, 9 Paediatric Cushing syndrome differs from adult-onset Cushing syndrome in aspects including effects on growth (weight gain with concomitant height deceleration), atypical physical presentation (such as lack of centripetal obesity or typical striae), delayed or suppressed puberty, and variable mental health problems and neurocognitive function deficits.10 Diagnosis of paediatric Cushing syndrome is therefore challenging, and delayed evaluation is common.
Research in context
Evidence before this study
Endogenous Cushing syndrome is a rare endocrine condition. Diagnosis can be challenging and delay treatment. We searched PubMed for articles published in English on paediatric Cushing syndrome using terms “Cushing” AND “children” from database inception to May 5, 2023. Although several case series of paediatric Cushing disease were identified, only a few studies of the various causes of paediatric endogenous Cushing syndrome were available.
Added value of this study
To our knowledge, this cohort of paediatric endogenous Cushing syndrome of various causes is one of the largest sources of cumulative clinical, anthropometric, and biochemical data on the presentation, diagnosis, and management. We confirm that baseline biochemical data cannot aid differential diagnosis of Cushing syndrome subtypes. However, evidence suggests that minimally invasive stimulation tests could be a safe alternative to interventional sampling procedures such as inferior petrosal sinus sampling. We provide the prevalence of complications related to Cushing syndrome. Long-term outcomes of paediatric patients with pituitary corticotroph tumours recurrence is possible up to 8 years after initial remission.
Implications of all the available evidence
Data from this large paediatric cohort inform the evaluation, diagnosis, and long-term care of patients with paediatric Cushing syndrome. We recommend an algorithm for the diagnosis of patients and screening of complications. Screening for recurrence in patients with Cushing disease is indicated for this age group, at least for the first decade after surgery.
We have evaluated a large cohort of children and adolescents with endogenous Cushing syndrome of various causes. The aim of the study was to document anthropometric, clinical, and biochemical characteristics, complications, and outcomes of paediatric endogenous Cushing syndrome to aid clinicians in the diagnosis and management of these patients.
Section snippets
Study design and participants
In this prospective, multisite cohort study, we screened participants who, from 1995 to 2023, had enrolled in studies under protocols 97-CH-0076 (clinicaltrials.gov, NCT00001595), 95-CH-0059 (NCT00001452), and 00-CH-0160 (NCT00005927) at the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD, Bethesda, MD, USA). Paediatric patients (18 years or younger at time of presentation) with a diagnosis of Cushing syndrome were eligible for inclusion in the study. We
Results
342 patients with paediatric Cushing syndrome were included in the study (table 1). 278 patients were referred from centres in the USA, and 64 patients were referred from centres outside of the USA. 261 (76%) patients were diagnosed with Cushing disease, 74 (22%) patients were diagnosed with adrenal-associated Cushing syndrome, and seven (2%) patients were diagnosed with ectopic Cushing syndrome. Patients with adrenal-associated Cushing syndrome were diagnosed at a younger age than patients
Discussion
We present extensive and unique data on presentation, diagnosis, and follow-up of paediatric patients with three diagnostic types of endogenous Cushing syndrome. Clinical and anthropometric characteristics were similar across subtypes of Cushing syndrome, but biochemical tests differed. We also present extensive information on complications; hypertension, insulin resistance, dyslipidaemia, and elevated ALT were common. Long-term follow-up of patients revealed excellent postoperative prognosis,
Data sharing
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Declaration of interests
CAS holds patents on the function of the PRKAR1A, PDE11A, and GPR101 genes and related issues; his laboratory had received research funding on GPR101, and on abnormal growth hormone secretion and its treatment by Pfizer. CAS receives support from ELPEN and has been consulting for Lundbeck Pharmaceuticals and Sync. CT reports receiving research funding on treatment of abnormal growth hormone secretion by Pfizer.
Cushing’s syndrome is a rare entity in children. Adrenal tumour is the common cause of this syndrome in young children, whereas, iatrogenic causes are more common among older children. We report a 4 year old male child diagnosed with Cushing syndrome due to a right adrenal adenoma; the child presented with obesity and increase distribution of body hair. After thorough investigation and control of hypertension and dyselectrolytemia, right adrenalectomy was performed. The patient had good clinical recovery with weight loss and biochemical resolution of Cushing’s syndrome.
1. Introduction
Cushing’s syndrome (CS) is rarely encountered in children. The overall incidence of Cushing syndrome is approximately 2–5 new cases per million people per year. Only approximately 10% of the new cases each year occur in children [1]. Unlike in adults, a male-to-female predominance have been observed in infants and young toddlers [[1], [2], [3]]. Although iatrogenic causes are common in children above seven years of age, adrenal causes (adenoma, carcinoma or hyperplasia) are common in children of younger age [4]. We report a 4 year old boy diagnosed with Cushing syndrome caused by a right adrenal adenoma, who had presented with obesity and increase distribution of body hair. Right adrenalectomy was performed and clinical stabilization resulted in weight loss and biochemical resolution of Cushing’s syndrome. (see Fig. 5)
2. Case report
A 4 years old boy presented with complaints of excessive weight gain of 5 months duration and increase frequency of micturition and appearance of body hair for 4 months. There was no history of any other illness, medication or steroid intake. The child was first born at term by normal vaginal delivery and birth weight of 3 kg. Physical examination revealed a chubby boy with moon face, buffalo hump, protruding abdomen, increase body hair and appearance of coarse pubic hair (Fig. 1). His intelligent quotient (IQ) was appropriate for his age and sex. His younger sibling was in good health and other family members did not have any metabolic or similar problems.
Fig. 1. The child with moon face, protruded abdomen and coarse body hair.
The patient’s body length was 92cm (between -2SD to -3SD), weight 20kg (between 1 SD and 2 SD), weight for height >3SD, and BMI was 23.6 (BMI for age >3 SD). His blood pressure on right arm in lying position was 138/76 mm Hg (above 99th percentile for height and age).
with a concurrent plasma ACTH level of < 5 pg/ml (n value < 46 pg/ml).
His serum cortisol following low dose dexamethasone suppression test (1mg dexamethasone at 11pm) at 8 am next morning was 22.1 μug/dl and his 24 hours urine catecholamine fraction was within normal limit.
Ultrasonography of abdomen revealed a heterogenous predominantly hypoechoic right supra renal mass. Contrast enhanced CT abdomen revealed well defined soft tissue density lesion (size −5.2 cm × 5.2 cm x 5.7cm) in right adrenal gland with calcifications and fat attenuations showing mild attenuation on post contrast study (Fig. 2).
Fig. 2. CECT shows right adrenal mass with calcification and mild attenuation on post-contrast study.
The child was started on oral amlodipine 2.5mg 12hourly; after 5days blood pressure became normal. For hypokalemia oral potassium was given @20 meq 8 hourly and serum potassium value became normal after 4 days. Right laparoscopic adrenalectomy was planned. but due to intra operative technical problems it was converted to an open adrenalectomy with right subcostal incision. A lobulated mass of size 9 cm × 5 cm x 4 cm with intact capsule was excised. The tumour weighed 230 gm. There was no adhesion with adjacent organs, three regional nodes were enlarged but without any tumour tissue. Inferior vena cava was spared. Histopathology report was consistent with adrenal adenoma (Fig. 3) (see Fig. 4).
Fig. 4. Microphotograph (100 × 10) showing intact capsule and adrenal tumour cells, which are larger in size with nuclear pleomorphism, inconspicuous nucleoli, cytoplasm of the tumour cells are abundant, eosinophilic and vacuolated.
Fig. 5. Physical appearance 4 months after adrenalectomy.
Post operative management: during post operative period hypokalemia and flaxuating blood sugar level was managed with oral potassium and oral glucose supplement. patient developed mild cough and respiratory distress on post op day 2, it was managed with salbutamol nebulization and respiratory physio therapy. Patient developed minor ssi and discharged on 10 th post operative day with oral prednisolone supplementation.
Follow up: the patient was followed up 2week after discharge and then every monthly, the oral prednisolone was gradually tapered and completely withdrawn on 2nd month after surgery.The patient experienced no post-surgical complications. After 4 months of surgery he reduces 6 kgs of his body weight with BMI of 16.5 (between median and 1SD) & BP 100/74 mm hg (within normal range), the moon face, buffalo hump, central obesity disappeared, morning 8am serum cortisol level was found within normal range 14 μg/dl (n value 6–23 μg/dl).
3. Discussion
Cushing’s syndrome is caused by prolonged exposure to supraphysiological levels of circulating glucocorticoids, which may be endogenously or exogenously derived. During infancy, CS is usually associated with McCune-Albright syndrome; adrenocortical tumours most commonly occur in children under four years of age and Cushing’s disease (ACTH dependent) is the commonest cause of CS after five years of age [5]. Primary adrenocortical tumours (ACTs) account for only 0.3–0.4% of all childhood neoplasms. Almost a third of these tumours manifests as Cushing syndrome and over 70% of the unilateral tumours in young children are often malignant [2,3,6,7]. There seems to be a bimodal incidence of these tumours, with one peak at under 5 years of age and the second one in the fourth or fifth decades of life. ACTs may be associated with other syndromes, such as, Li-Fraumeni syndrome, Beckwith-wiedemann syndrome, isolated hemihypertrophy, or even a germline point mutation of P53 tumour suppressor gene as reported in a series from Brazil [8]. In comparison to adult CS, growth failure with associated weight gain is one of the most reliable indicators of hypercortisolaemia in pediatric CS. The parents often fail to notice facial changes and growth failure and hence the diagnosis is often delayed. In one study, the mean time from appearing symptoms to diagnosis in 33 children with Cushing’s disease was 2.5 years [5]. More recently the comparison of height and BMI SDS measurements provided a sensitive diagnostic discriminator in pediatric patients with CD and those with simple obesity [9]. In the present case, the parents observed noticeable changes in his face and presence of body hair, which made them to bring the child to medical attention. A review of 254 children on the International Pediatric Adrenocortical Tumour Registry identified virilization as the most common manifestation [10]. About 10% of the tumours can be non-functional at presentation, and approximately one third of pediatric patients present with hypertension. Majority of patients (192/254) in the Registry had localized disease and metastatic disease was found in less than 5% of cases. Older children with CS or mixed androgen and cortisol secreting adrenocortical tumours had a worse prognosis compared to younger children [10]. The present case had mild hypertension as well as dyselectrolytemia at presentation, which could be controlled with medication. He had a single adenoma confined to the adrenal gland and there was no evidence of malignancy. After surgical excision of the tumour and the right adrenal gland, the patient made rapid improvement in clinical condition and has been on follow up for last 7 months.
4. Conclusion
Pediatric adrenocortical tumours (ACTs) are most commonly encountered in females and in children less than four years. But our case being an 4-year-old boy forms a rare presentation of endogenous Cushing’s syndrome due to adrenal adenoma. Cushing’s syndrome in this child was controlled after right adrenalectomy.
Patient consent
Informed written consent was taken.
Funding
No funding or grant support.
Authorship
All authors attest that they meet the current ICMJE criteria for authorship.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The treatment of adrenal insufficiency with hydrocortisone granules in children with congenital adrenal hyperplasia (CAH) was associated with an absence of adrenal crises and normal growth patterns over a 2-year period, according to study findings published in The Journal of Clinical Endocrinology and Metabolism.
The study included a total of 17 children with CAH and 1 child with hypopituitarism. All included participants were <6 years old who were receiving current adrenocortical replacement therapy, including hydrocortisone with or without fludrocortisone. Hydrocortisone medications used in this population were converted from pharmacy compounded capsules to hydrocortisone granules without changing the dose.
These study participants were followed by study investigators for 2 years. Glucocorticoid replacement therapy was given three times a day for a median treatment duration of 795 days. Treatment was adjusted by 3 monthly 17-hydroxyprogesterone (17-OHP) profiles in children with CAH.
There were a 150 follow-up visits throughout the study. At each visit, participants underwent assessments that measured hydrocortisone dose, height, weight, pubertal status, adverse events, and incidence of adrenal crisis.
A total of 40 follow-up visits had changes in hydrocortisone doses based on salivary measurements (n=32) and serum 17-OHP levels (n=8).
At time of study entry, the median daily doses of hydrocortisone were 11.9 mg/m2 for children between the ages of 2 to 8 years, 9.9 mg/m2 for children between 1 month and 2 years, and 12.0 mg/m2 for children <28 days of age. At the end of the study, the respective doses for the 3 age groups were 10.2, 9.8, and 8.6.
The investigators observed no trends in either accelerated growth or reduced growth; however, 1 patient with congenital renal hypoplasia and CAH did show reduced growth. While 193 treatment-emergent adverse events, including pyrexia, gastroenteritis, and viral upper respiratory tract infection, were reported in 14 patients, there were no observed adrenal crises.
Limitations of this study included the small sample size as well as the relatively high drop-out rate of the initial sample.
The researchers concluded that “hydrocortisone granules are an effective treatment for childhood adrenal insufficiency providing the ability to accurately prescribe pediatric appropriate doses.”
Disclosure: Several study authors declared affiliations with the pharmaceutical industry. Please see the original reference for a full list of authors’ disclosures.
RSS Error: A feed could not be found at `http://www.oconnormusic.org/feed.xml`; the status code is `503` and content-type is `text/html; charset=us-ascii`
RSS Error: A feed could not be found at `http://www.oconnormusic.org/bios.xml`; the status code is `503` and content-type is `text/html; charset=us-ascii`