Abstract
Introduction and importance
Pheochromocytoma and Cushing’s syndrome are rare endocrine conditions caused by tumors in the adrenal gland. These conditions are classified under Multiple Endocrine Neoplasia (MEN) syndrome, characterized by the development of multiple tumors in the endocrine system. However, diagnosing these conditions can be challenging as they often lack clear symptoms, requiring careful evaluation, monitoring, and treatment to prevent complications.
Case presentation
A 23-year-old male recently presented with right-sided abdominal fullness and lipoma-like masses on the torso. Over a span of six months, the abdominal mass nearly doubled in size, accompanied by elevated levels of catecholamines, cortisol, parathyroid hormone (PTH), and calcitonin. Surprisingly, the patient remained asymptomatic despite these abnormal lab values. CT imaging revealed a substantial increase in the size of the mass in the right adrenal gland, from 6 × 7 cm to approximately 11.2 × 10.2 × 9 cm.
Clinical discussion
Pheochromocytoma secretes catecholamines and often leads to hypertension and related symptoms. Interestingly, most individuals with pheochromocytoma do not exhibit obvious symptoms, necessitating blood and urine tests, along with imaging studies, for accurate diagnosis. The size of the tumor does not necessarily indicate the severity of symptoms. MEN-2, a genetic syndrome, is characterized by pheochromocytoma, medullary thyroid carcinoma, and hyperparathyroidism. Additionally, methods for diagnosing Cushing’s syndrome, caused by excess cortisol production, are discussed.
Conclusion
Early diagnosis and genetic counseling are crucial in preventing complications associated with these conditions. By identifying them, appropriate treatment can be ensured for positive outcomes of patients and their families.
Keywords
Abbreviations
- CT
- MRI
-
Magnetic resonance imaging
- USG
- 131I-MIBG
-
iodine 131 labeled meta-iodobenzylganidine
- RAAS
-
Renin-angiotensin-aldosterone system
1. Introduction
Pheochromocytoma are catecholamine secreting tumors of chromaffin cells of adrenal medulla. It can be found anywhere in the body, with the majority being intra-abdominal and those other than adrenal medulla are referred to as paragangliomas [1,2]. Pheochromocytoma typically secretes norepinephrine and epinephrine, with norepinephrine being the primary catecholamine. However, some tumors may only secrete one of the two, and rarely, some may secrete dopamine or dopa [3].
Vast majority >90 % of adrenal neoplasms are benign non-functional adenomas [4].About 10 % of pheochromocytomas are malignant and 10 % of cases are found on both sides. Additionally, approximately 40 % of pheochromocytomas are caused by genetic factors and can be associated with inherited syndromes [5].
Pheochromocytoma is found to be associated with MEN-2. MEN-2 is a hereditary genetic condition that is caused by a de novo mutation in the RET gene. It is inherited in an autosomal dominant fashion and is mainly characterized by medullary thyroid carcinoma, pheochromocytoma and parathyroid adenoma or hyperplasia [6].
MEN syndrome can be MEN-1, MEN-2A and MEN-2B. MEN-1 is characterized by pituitary tumors (prolactin or growth hormone), pancreatic endocrine tumors and parathyroid adenomas. Additionally, other tumors such as foregut carcinoids, adrenocortical adenomas, meningioma, lipomas, angiofibromas and collagenomas may also occur in MEN-1. MEN-2A is characterized by medullary thyroid carcinoma, pheochromocytoma, and parathyroid adenoma/hyperplasia; it can also be associated with cutaneous lichen amyloidosis and Hirschsprung disease. On the other hand, MEN-2B is characterized by familial medullary thyroid cancer, pheochromocytoma, mucosal neuromas, gastrointestinal tract issues, musculoskeletal and spinal problems. [7].
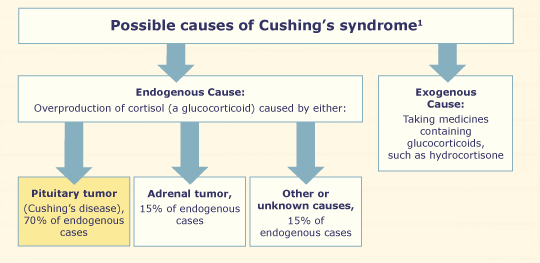
Cushing syndrome results from hypercortisolism and is characterized by hypertension, weight gain, easy bruising, and central obesity [4]. Cushing’s disease refers to ACTH-dependent cortisol excess caused by a pituitary adenoma, while ACTH-independent cortisol excess due to non-pituitary causes such as excess use of glucocorticoids, adrenal adenoma, hyperplasia, or carcinoma is referred to as Cushing syndrome [8].
This case report has been written according to the SCARE checklist [9].
2. Case presentation
A 23-year-old male presented to our surgery department with the chief complaint of right sided abdominal fullness for six months. According to the patient a mass was incidentally reported six months back while he was under-evaluation for mild trauma due to road traffic accident. Six months back, the mass was approximately 6 × 7 cm, while at the time of presentation to our department the mass was approximately 11.2 × 10.2 × 9 cm (CT abdomen) which was globular in shape, had regular margin, and moved with respiration. He had no history of hypertension, headache, palpitation, sweating, pallor, recent weight loss, abdominal pain, psychological disturbance, dizziness, loss of consciousness, dark color urine, burning micturition, had normal bowel and bladder habit.
Past history and family history were insignificant. He was not under any long-term medication and no known drug allergies. He occasionally smokes and consumes alcohol.
On physical examination at the time of presentation, multiple soft, mobile, painless, subcutaneous nodules like lipoma were present over the torso. His height was 176.8 cm, weight 68 kg, BMI 21.8 kg/m2 (body mass index). He had blood pressure of 110/70 mm of Hg taken in left arm at sitting position, heart rate of 62 beats/min, respiratory rate of 24/min, temperature of 96.6 °F, SPO2 of 98 % at right hand. A mass was palpable on the right side of abdomen, otherwise abdomen was soft, non-tender, normal bowel sound was present. Chest, cardiac and neurologic examinations were all normal.
Initial laboratory evaluation revealed 24 h. urine metanephrine of 5415 μg/24 h (normal: 25–312 μg/24 h.); 24 h. urine VMA of 32.2 mg/24 h. (normal: <13.60 mg/24 h.); serum cortisol of 535.16 nmol/l after overnight low dose dexamethasone(1 mg) suppression test (normal: <50 nmol/l);24 h. Urine free cortisol of 526.61 nmol/24 h. (normal: 30–145 nmol/24 h) PTH(intact) of 89.2 pg./ml (normal: 15–65 pg./ml); serum calcitonin of 15.2 pg./ml (normal: ≤8.4 pg./ml); serum CEA of 4.72 ng/ml (normal: 0.0–4.4 ng/ml); serum DHEA of 1.19 ng/ml (normal: 1.7–6.1 ng/ml). Baseline investigation: Hematology, urine routine/microscopic, electrolytes were within the normal range.
Additional laboratory findings were as in the Table 1.
Table 1.
| Lab evaluation | Result | Reference | Unit |
|---|---|---|---|
| Metanephrine, urine 24 h | 5415 | 25–312 | μg/24 h |
| VMA, urine 24 h | 32.2 | <13.60 | mg/24 h |
| VMA, urine | 12.88 | – | ng/l |
| Cortisol, serum, overnight DST | 535.16 | <50 | nmol/l |
| Cortisol, urine 24 h | 526.61 | 30–145 | nmol/24 h |
| ACTH, complete | 28.3 | 7.2–63.3 | pg/ml |
| DHEA, serum | 1.19 | 1.7–6.1 | ng/ml |
| CEA, serum | 4.72 | 0.0–4.4 | ng/ml |
| Phosphorus, serum | 3.0 | 2.5–4.5 | mg/dl |
| Albumin, serum | 5.2 | 3.5–5.2 | g/dl |
| Calcitonin, serum | 15.2 | ≤8.4 | pg/ml |
| Calcium, serum | 8.94 | 8.6–10.0 | mg/dl |
| PTH (intact) | 89.2 | 15–65 | pg/ml |
| aldosterone | 8.7 | 7.0–30 | g/dl |
| Plasma rennin activity | 1.42 | 0.10–6.56 | ng/ml/h |
| Aldosterone-rennin ratio | 6.13 | ≤20 | |
| Creatinine, urine | 36 | – | mg/dl |
DST – dexamethasone suppression test; VMA – vanilmandelic acid; ACTH – adrenocorticotropic hormone; DHEA – dehydroepiandrosterone; CEA – carcino-embryonic-antigen; PTH – parathyroid hormone.
2.1. USG abdomen
USG abdomen (Fig. 1, Fig. 2) showed well defined mixed echoic area in Right adrenal region measuring 12.7 × 10.7 cm in size. There was presence of internal vascularity with multiple foci of cystic compound. The lesion displaced the right kidney inferiorly.

Fig. 1. USG abdomen.

Fig. 2. USG abdomen.
2.2. Plane and contrast CT scan of abdomen
Plane and contrast CT scan of Abdomen (Fig. 3) showed approximately 11.2 × 10.2 × 9 cm sized, relatively well defined heterogeneous soft tissue density lesion with well-defined enhancing wall in right adrenal region. Non-enhancing areas were noted within the mass suggestive of necrosis. Few calcific foci were noted within the mass with no obvious hemorrhagic component. The lesion showed heterogeneous enhancement post contrast image.

Fig. 3. CT abdomen.
After all the workup patient was given diagnosis of right sided Pheochromocytoma associated with MEN syndrome, with ACTH-independent Cushing’s syndrome and right adrenalectomy was performed.
2.3. Pathology report
2.3.1. Gross descriptions
The specimen was globular mass measuring 14.5 × 10 cm, with smooth outer surface. On sectioning, the mass was well circumscribed, soft and yellow-brown, predominantly solid with cyst formation. The size of cyst ranges from 0.3 to 3.5 cm in diameter. Areas of hemorrhages were noted.
2.3.2. Microscopic description
Section showed tumor cells arranged in well-defined nests (Zellballen), alveolar and diffuse pattern with intervening fibrovascular stroma. The cells were intermediate to large sized, polygonal with finely granular amphophilic cytoplasm. The nuclei showed mild to moderate pleomorphism and were round to ovoid, with prominent nuclei noted. No capsular invasion, vascular invasion and necrosis. Areas of hemorrhage were seen. Mitosis 0–1/10 high power field was noted (Figs. 4 and 5).

Fig.a Diffuse Zellbalen pattern with intervening fibrous stroma.
Fig.b Mild to moderate pleomorphic nuclei with abundant hemorrhage.
Fig.c Low power field with intact capsule.

Figs. 4 and 5. Fig. 4 Intra-operative resection of tumor; Fig. 5 tumor after resection.
3. Discussion
In Pheochromocytoma activation of the alpha-one adrenergic receptor by catecholamine in the vascular bed causes vasoconstriction and leads to a rise in blood pressure. Similarly, activation of the beta-one receptor in the heart enhances the chronotropic and inotropic effect of the myocardium, leading to an increase in heart rate and cardiac output. In addition, activation of the beta-one receptor in the juxtaglomerular cells of the kidney activates the RAAS system. These receptor activation result in cardiovascular and sympathetic changes, such as hypertension, palpitation, headache, sweating, trembling, and anxiety [10].
In Pheochromocytoma, the patient may have a 10-fold increase in plasma catecholamines, but the hemodynamic response can still fall within the normal range due to desensitization of the cardiovascular system. When catecholamine levels are elevated for a prolonged period, the alpha-one receptors in blood vessels may be down-regulated, making norepinephrine unresponsive in raising peripheral vascular resistance, which can lead to normal blood pressure. Similarly, a marked decrease in beta-one receptors in the heart could explain the normal heart rate, which was observed in our asymptomatic patient with Pheochromocytoma [11].
Sometimes in asymptomatic patients, the size of the tumor tends to be larger than in those with hyperfunctioning tumors [12]. However, medical interventions such as surgery, anesthesia induction, intravenous urography contrast, or manipulation of the tumor can trigger adrenergic and hypertensive crises, so biopsy is usually contraindicated in pheochromocytoma [13].
The diagnosis of pheochromocytoma is typically based on measuring plasma and urinary levels of catecholamines and their derivatives such as metanephrine and vanillylmandelic acid. The most reliable test is the measurement of urinary metanephrine as its excretion levels are relatively higher [13,14]. The combination of 131I-MIBG scintigraphy along with diagnostic urinary and blood tests can further enhance the sensitivity of the test. Specifically, the urinary normetanephrine test is considered the most sensitive single test for detecting Pheochromocytoma [15,16].
In addition to a 24-h urine test and blood test, if the lab results are positive for Pheochromocytoma or paragangliomas, further diagnostic tests may be recommended, such as a CT scan, MRI, m-iodobenzylganidine (MIBG) imaging, or positron emission tomography (PET) [16,17]. In our patient 24 h. urine metanephrine of 5415 μg/24 h (normal: 25–312 μg/24 h.); 24 h. urine VMA of 32.2 mg/24 h. (normal: <13.60 mg/24 h.) and imaging confirmation of right adrenal mass lead to the diagnosis of right sided pheochromocytoma.
Our patient with pheochromocytoma was tested for parathyroid hormone and calcitonin due to the association of pheochromocytoma with MEN-2 [18]. MEN-2 can be diagnosed biochemically by measuring the baseline levels of calcitonin, parathyroid hormone and serum calcium along with blood tests for catecholamines and their metabolites to detect pheochromocytoma [19]. In our patient, multiple soft, mobile, painless, subcutaneous nodules like lipoma were present over the torso(MEN-1) and high levels of parathyroid hormone and calcitonin were detected(MEN-2). These findings can be correlated with MEN syndrome.
USG of the neck revealed no abnormalities of thyroid and parathyroid gland in our patient so prophylactic thyroidectomy was not done, instead he was counseled for follow up if any symptoms or thyroid swelling appears.
The diagnosis of Cushing’s syndrome typically involves measuring the levels of 24-h urine free cortisol and assessing the suppression of cortisol in response to a 1 mg overnight dexamethasone test. If cortisol levels remain elevated despite the test, the next step is to measure serum ACTH levels. If ACTH levels are suppressed, it suggests an ACTH-independent cause of Cushing’s syndrome, while elevated ACTH levels suggest an ACTH-dependent cause. Further evaluation may include a CT scan of the chest, abdomen, and pelvis to identify potential ectopic sources, as well as an MRI of the pituitary gland [8]. Our patient had a high level of 24 h. urine free cortisol of 526.61 nmol/24 h (reference range: 30–145 nmol/24 h) and serum cortisol of 535.16 nmol/L(reference range: <50 nmol/L) after overnight 1 mg dexamethasone suppression test, but normal level of ACTH of 28.3 pg./ml (reference range: 7.2–63.1 ng/ml), this suggests the diagnosis of ACTH independent Cushing’s syndrome.
4. Conclusion
Large Pheochromocytoma patients can be asymptomatic and can present in association with other endocrine disorders. So proper evaluation is necessary to find out associated conditions and manage accordingly to prevent the possible outcomes.
Patient consent
Written, informed consent was obtained from the patient for the publication of the report.
Ethical approval
It is exempted at my institution. We don’t need to take approval from ethical committee for case report.
Funding
N/A.
Author contribution
Conceptualization: Sanjit Kumar Shah.
Clinical diagnosis and patient management: Mahipendra Tiwari.
Microscopic slide preparation: Sneh Acharya.
Writing original draft: Sanjit Kumar Shah and Avish Shah.
All authors were involved in reviewing, editing, supervision and in preparing the final
manuscript.
Guarantor
Guarantor: Sanjit Kumar Shah
Email: sanjitshah023@gmail.com
Conflict of interest statement
N/A.
References
- [1]
P.J. Klingler, T.P. Fox, D.M. Menke, J.M. Knudsen, J.T. FulmerPheochromocytoma in an incidentally discovered asymptomatic cystic adrenal massMayo Clin. Proc., 75 (5) (2000), pp. 517-520, 10.4065/75.5.517
- [2]
K. Salmenkivi, J. Arola, R. Voutilainen, et al.Inhibin/activin betaB-subunit expression in pheochromocytomas favors benign diagnosisJ. Clin. Endocrinol. Metab., 86 (5) (2001), pp. 2231-2235, 10.1210/jcem.86.5.7446
- [3]
W.M. Manger, R.W. GiffordPheochromocytomaJ. Clin. Hypertens (Greenwich)., 4 (1) (2002), pp. 62-72, 10.1111/j.1524-6175.2002.01452.x
This article is free to access.
- [4]
S.R. Pingle, F. Jalil, D. Millar, C.D. Malchoff, B.T. RistauIsolated DHEAS production by an adrenal neoplasm: clinical, biochemical and pathologic characteristicsUrol. Case Rep., 31 (2020), Article 101148Published 2020 Feb 26Published 2020 Feb 26
- [5]
I. Jandou, A. Moataz, M. Dakir, A. Debbagh, R. AboutaiebMalignant pheochromocytoma: a diagnostic and therapeutic dilemmaInt. J. Surg. Case Rep., 83 (2021), Article 106009, 10.1016/j.ijscr.2021.106009
- [6]
L. Canu, G. Parenti, G. De Filpo, M. MannelliPheochromocytomas and Paragangliomas as causes of endocrine hypertensionFront. Endocrinol. (Lausanne)., 10 (2019), p. 333Published 2019 Jun 4Published 2019 Jun 4
- [7]
G.G. Callender, T.A. Rich, N.D. PerrierMultiple endocrine neoplasia syndromesSurg. Clin. North Am., 88 (4) (2008), p. 863viiiviii
- [8]
R. Pivonello, M. De Leo, A. Cozzolino, A. ColaoThe treatment of Cushing’s diseaseEndocr. Rev., 36 (4) (2015), pp. 385-486, 10.1210/er.2013-1048
- [9]
R.A. Agha, T. Franchi, C. Sohrab, G. Mathew, A. Kirwan, A. Thomas, et al.The SCARE 2020 guideline: updating consensus Surgical Case Report (SCARE) guidelinesInt. J. Surg., 84 (1) (2020), pp. 226-230
- [10]
L. Canu, G. Parenti, G. De Filpo, M. MannelliPheochromocytomas and Paragangliomas as causes of endocrine hypertensionFront. Endocrinol. (Lausanne)., 10 (2019), p. 333Published 2019 Jun 4Published 2019 Jun 4
- [11]
E. Bravo, F. Fouad-Tarazi, G. Rossi, et al.A reevaluation of the hemodynamics of pheochromocytomaHypertension., 15 (2 Suppl) (1990), pp. I128-I131, 10.1161/01.hyp.15.2_suppl.i128
- [12]
M.A. Blake, S.K. Krishnamoorthy, G.W. Boland, et al.Low-density pheochromocytoma on CT: a mimicker of adrenal adenomaAJR Am. J. Roentgenol., 181 (6) (2003), pp. 1663-1668, 10.2214/ajr.181.6.1811663
- [13]
R. Yu, A. Pitts, M. WeiSmall pheochromocytomas: significance, diagnosis, and outcomeJ. Clin. Hypertens (Greenwich)., 14 (5) (2012), pp. 307-315, 10.1111/j.1751-7176.2012.00604.x
This article is free to access.
- [14]
F. Zoghbi, C. Landault, N. Salmon, J.C. LegrandLes catécholamines et leurs dérivés dans l’exploration du phéochromocytome [Catecholamines and their derivatives in the study of pheochromocytoma]Ann. Med. Interne (Paris)., 134 (3) (1983), pp. 230-232
- [15]
U. Guller, J. Turek, S. Eubanks, E.R. Delong, D. Oertli, J.M. FeldmanDetecting pheochromocytoma: defining the most sensitive testAnn. Surg., 243 (1) (2006), pp. 102-107, 10.1097/01.sla.0000193833.51108.24
- [16]
G. Eisenhofer, M. PeitzschLaboratory evaluation of pheochromocytoma and paragangliomaClin. Chem., 60 (12) (2014), pp. 1486-1499, 10.1373/clinchem.2014.224832
This article is free to access.
- [17]
H.P. Neumann, D.P. Berger, G. Sigmund, et al.Pheochromocytomas, multiple endocrine neoplasia type 2, and von Hippel-Lindau disease [published correction appears in N Engl J Med 1994 Dec 1;331(22):1535]N. Engl. J. Med., 329 (21) (1993), pp. 1531-1538, 10.1056/NEJM199311183292103
- [18]
V. Amodru, D. Taieb, C. Guerin, et al.MEN2-related pheochromocytoma: current state of knowledge, specific characteristics in MEN2B, and perspectives [published correction appears in endocrine. 2020 Jun 2;:] [published correction appears in endocrine. 2020 Jul 14;:]Endocrine, 69 (3) (2020), pp. 496-503, 10.1007/s12020-020-02332-2
- [19]
C.J. Lips, J.W. Höppener, B.P. Van Nesselrooij, R.B. Van der LuijtCounselling in multiple endocrine neoplasia syndromes: from individual experience to general guidelinesJ. Intern. Med., 257 (1) (2005), pp. 69-77, 10.1111/j.1365-2796.2004.01429.x
This article is free to access.
Filed under: adrenal, Cushing's, Rare Diseases, symptoms | Tagged: Adrenal gland, MEN1, pheochromocytoma, subclinical Cushing's syndrome | Leave a comment »