Introduction
Pasireotide (PAS) is a novel somatostatin receptor ligand (SRL) with a high affinity for the somatostatin receptor (SSR) type 5 (1, 2). Somatotroph adenomas are usually responsive to first-generation SRLs (octreotide and lanreotide), as they are able to reduce growth hormone (GH) secretion through SSR type 2 (3). In the flow-chart of acromegaly treatment, PAS is suggested in case of resistance to first-generation SRLs, as SSR type 5 is also abundantly expressed in GH-secreting adenomas (3). A phase III study with PAS long-acting release (LAR) proved its efficacy in first-generation SRLs-resistant acromegalic patients after 6 months (4). In the extension study (Colao A et al.), 37% of patients achieved normalization of insulin-like growth factor 1 (IGF-1) and/or GH levels <1 µg/L, considering both those performing the extension treatment and those crossing over from the first-generation SRL-control group to the PAS LAR group. Nearly two-thirds of responses were achieved after at least 6 months of treatment. Up-titration of the dose from 40 to 60 mg monthly enriched the number of responders, suggesting that the PAS LAR effect may be both time- and dose-dependent (5). Concomitant improvement in signs and symptoms has also been confirmed in other series (6–9).
SSR type 5 is the predominant isoform in human corticotroph adenomas, since it is not down-regulated by high cortisol levels, as SSR type 2 does. Therefore, PAS is the only SRL available in patients with Cushing’s Disease (CD) (2). In a phase III study, subcutaneous (s.c.) PAS proved to be effective in normalizing urinary free cortisol (respectively in 13% and 25% of patients taking 600 µg or 900 µg bis-in-die for 12 months) (10), achieving significant clinical improvement (11). In the same clinical setting, PAS LAR showed similar efficacy and safety profiles (12). These benefits could be maintained for up to 5 years in an extension study (13, 14). In a recent meta-analysis, PAS treatment provided disease control in 44% of 522 patients with CD (15). Patients harbouring USP-8 mutations demonstrated an increased SSR type 5 expression in the corticotroph adenoma, increasing the likelihood of a positive response to PAS therapy (16). The safety profile of PAS is similar to that of first-generation SRLs, except for a significant worsening in glucose homeostasis (17).
Despite the normalization of hormonal excess, another goal of the medical treatment in GH-secreting pituitary adenomas is the reduction of the size of the adenoma (18). First-generation SRLs proved to be effective in achieving tumor shrinkage in acromegaly: Endocrine Society clinical practice guidelines suggested their role as primary therapy in poor surgical candidates and in those with extrasellar extension without chiasmal compression (18). Cozzi et al. reported in a large prospective cohort of acromegalic patients a significant Octreotide-induced tumor shrinkage in 82% of those receiving SRL as first-line treatment; most of them exhibited an early shrinkage with a progressive trend in reduction later on (19). A meta-analysis of 41 studies reported a significant tumor shrinkage in 50% of included patients (20). Data from the primary treatment with once-monthly lanreotide in surgical naïve patients demonstrated its efficacy in reducing tumor volume, achieving significant tumor shrinkage in 63% of them (21). Hypo-intensity on T2-weighted sequences at baseline magnetic resonance imaging (MRI) seems to predict tumor volume reduction during first-generation SRLs treatment (22). Regarding patients with CD, most patients presented a microadenoma, usually not aggressive or invasive: only in selected cases tumor shrinkage is an aim to achieve in patients with corticotropinoma.
As available data are scarce (or limited to selected studies), and the issue of pituitary adenoma shrinkage is of primary importance in the management of tumors that cannot be addressed through surgery, the aim of this systematic review and meta-analysis is to summarize available data regarding the effect of PAS on tumor size.
Materials and Methods
We used the Population-Intervention-Comparison-Outcome (PICO) model to formulate the research questions for the systematic review (23), as summarised in Figure 1. The systematic review and meta-analysis were conducted and are reported according to the Preferred Reporting Items for Systematic Reviews and Meta-Analysis of Diagnostic Test Accuracy Studies (PRISMA-DTA) statement (24). We registered the protocol on the International Prospective Register of Systematic Reviews database (PROSPERO, https://www.crd.york.ac.uk/PROSPERO, number CRD42022328152).
Figure 1
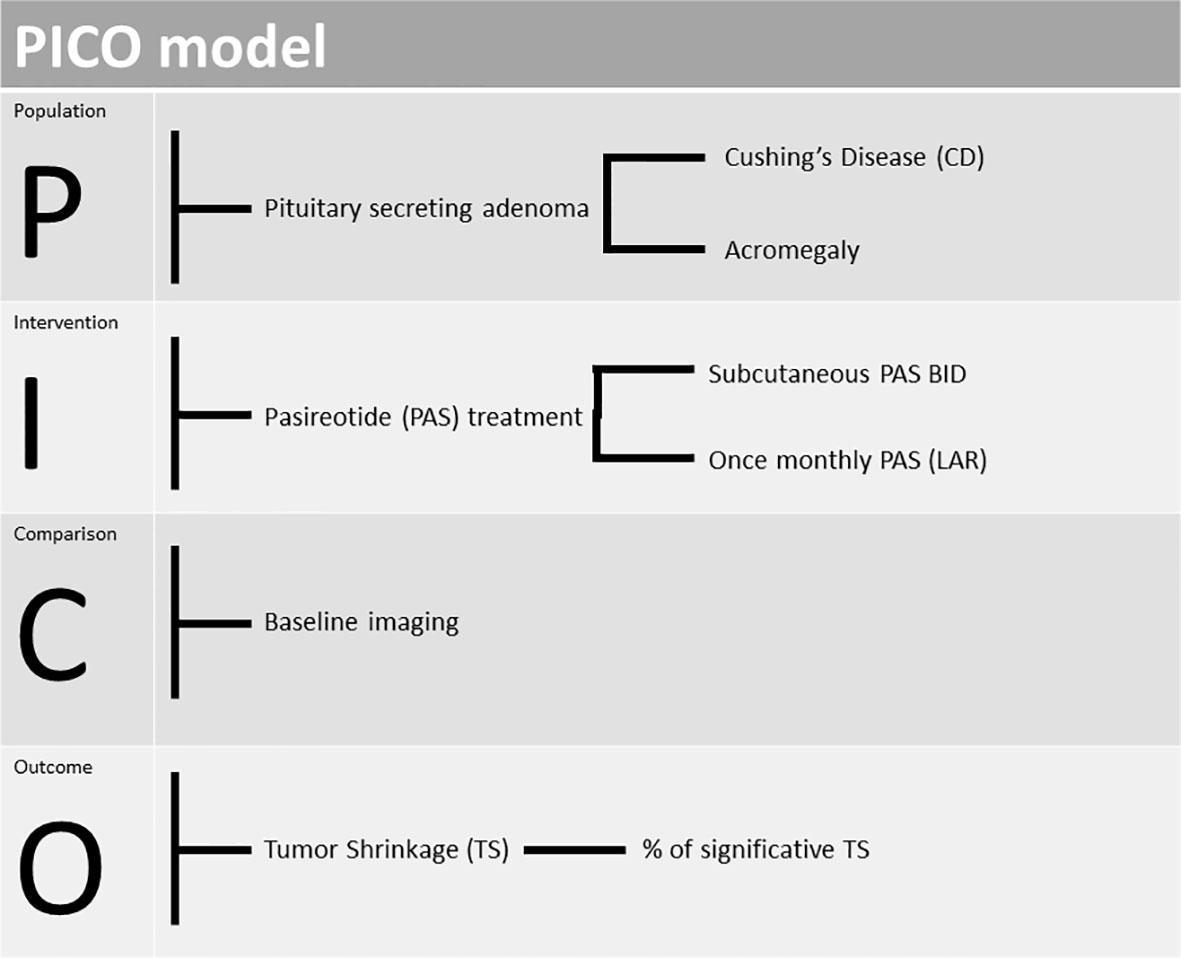 FIGURE 1
FIGURE 1 PICO (Population-Intervention-Comparison-Outcome) model design to our study.
Search Strategy
An extensive Medline search was performed for the research question by two of the authors (F.C. and A.M.) independently, discrepancies were resolved by discussion. The literature search was performed up to January 2022, no language restriction was applied. Research included the following keywords: 1) (“acromegalies” [All Fields] OR “acromegaly”[MeSH Terms] OR “acromegaly”[All Fields]) AND (“pasireotide”[Supplementary Concept] OR “pasireotide”[All Fields]); 2) (“pituitary ACTH hypersecretion”[MeSH Terms] OR (“pituitary”[All Fields] AND “ACTH”[All Fields] AND “hypersecretion”[All Fields]) OR “pituitary ACTH hypersecretion”[All Fields]) AND (“pasireotide”[Supplementary Concept] OR “pasireotide”[All Fields]).
Inclusion and exclusion criteria were specified in advance and protocol-defined, in order to avoid methodological bias for post-hoc analysis. The searches were designed to select all types of studies (retrospective, observational, controlled, randomized, and non-randomized) conducted in patients with acromegaly or CD treated with PAS as monotherapy; the assessment of the proportion of significant tumor shrinkage was an inclusion criterion. Search terms were linked to Medical Subject Headings when possible. Keywords and free words were used simultaneously. Additional articles were identified with manual searches and included a thorough review of other meta-analyses, review articles, and relevant references. Consolidation of studies was performed with Mendeley Desktop 1.19.8.
Study Selection
We included all original research studies conducted in adult patients that underwent PAS treatment used as monotherapy (s.c. bis-in-die and intramuscular once/monthly), that provided sufficient information about tumor size reduction during treatment. In case of overlapping cohorts of patients (as clinical trials with core and extension phases), we included only the extension study, in order to select those patients with measurable tumor shrinkage after long-term treatment. Local reports regarding patients involved in multicenter trials were excluded from the analysis, as they had been already considered in the larger series. Reviewers were not blinded to the authors or journals when screening articles.
Data Extraction and Quality Assessment
Two authors (F.C. and A.M.) read the included papers and extracted independently relevant data, any disagreements were resolved by discussion. If data were not clear from the original manuscript, the authors of the primary study were contacted to clarify the doubts.
Contents of data extraction in the selected paper included: name of the first author, year of publication, setting (referral centre, academic hospital, mono- or multi-centric collection), type of treatment, its dose schedule and duration, pituitary imaging method during follow- up, the endpoint type regarding adenoma size analysis (i.e. primary vs exploratory). When data were reported for each patient or for subgroups, a global percentage of significant tumor shrinkage was calculated considering all subjects involved in the study.
To assess the risk of bias in the included studies, the critical appraisal tool from Joanna Briggs’s Institute (JBI) was adapted to evaluate those considered in our metanalysis (25). Among the items proposed, we selected the most appropriate to our setting: 1. Were the inclusion criteria clearly identified? 2. Were diagnostic criteria for acromegaly or CD well defined? 3. Were valid methods applied to evaluate tumor shrinkage? 4. Was the inclusion of participants consecutive and complete? 5. Was the reporting of baseline participants’ features (demographic and clinical) complete? 6. Was the report of the outcomes clear? 7. Was the report of demographics of the involved sites complete? 8. Was statistical analysis appropriate? For each aspect we assigned as possible choices of answer: yes, no or unclear.
Data Synthesis and Analysis
A qualitative synthesis was performed summarizing the study design and population characteristics (age, male to female ratio, macro- to micro-adenoma ratio, prior treatments).
A random-effect model was used to estimate pooled effects. Forest plots for percentages of significant tumor size reduction were generated to visualize heterogeneity among the studies. In order to assess publication bias, despite the low number of articles considered, we performed funnel plot and asymmetry analysis adjusted for the low number of studies (an Egger’s regression test and a three-parameter selection model where two tailed p < 0.05 was considered statistically significant). The I2 test was conducted to analyze the heterogeneity between studies: an I2 >50% indicated a between-study heterogeneity.
Statistical analyses were performed with R: R-4.1.2 for Windows 10 (32/64 bit) released 2021-11-01 and R studio desktop RStudio Desktop 1.4.1717 for Windows 10 64 bit (R Foundation for Statistical Computing, Vienna, Austria, URL https://www.R-project.org/).
Results
Study Selection
The study selection process for acromegaly is depicted in Figure 2. The electronic search revealed 179 articles, with one duplicate (N = 178). After the first screening, 141 articles did not meet the eligibility criteria and were discarded. The full-text examination of the remaining studies excluded additional 31 articles: 27 did not provide adequate data about tumor size, two represented the core phase of an extension study, another one referred to a subset of patients from a larger study, and the last one did not provide sufficient data about the percentage of tumor size reduction. Thus, six studies fulfilling eligibility criteria (reported in Tables 1, 2), were selected for data extraction and analysis.
Figure 2
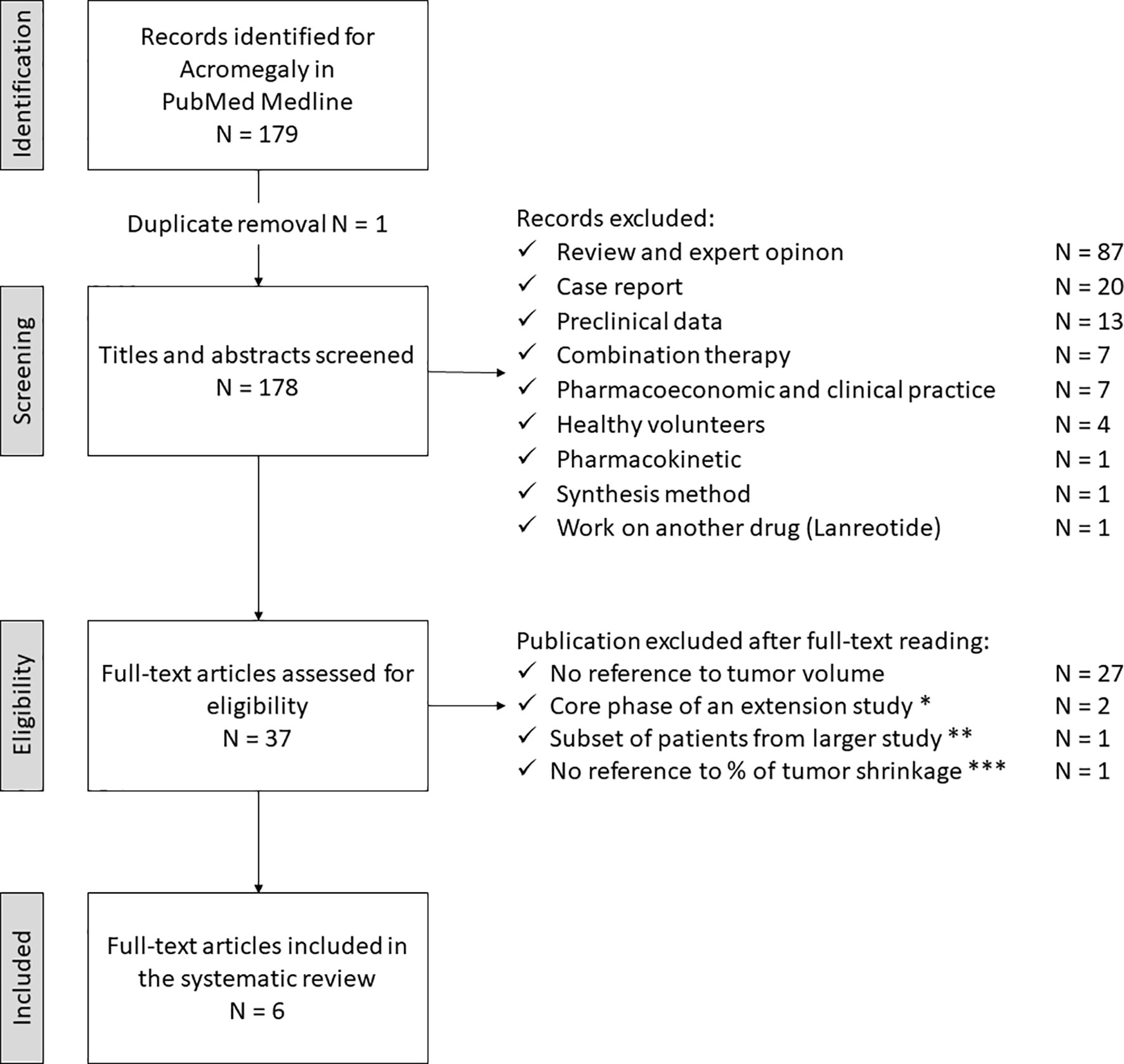 FIGURE 2
FIGURE 2 Search strategy for acromegaly. * Petersenn S, 2010 (PAS sc, phase II) and Colao A, 2014 (PAS LAR). ** Shimon I, 2015 (PAS LAR). *** Tahara S, 2019 (PAS LAR, phase II). PAS, pasireotide, sc, subcutaneous, LAR, long-acting release.
Table 1
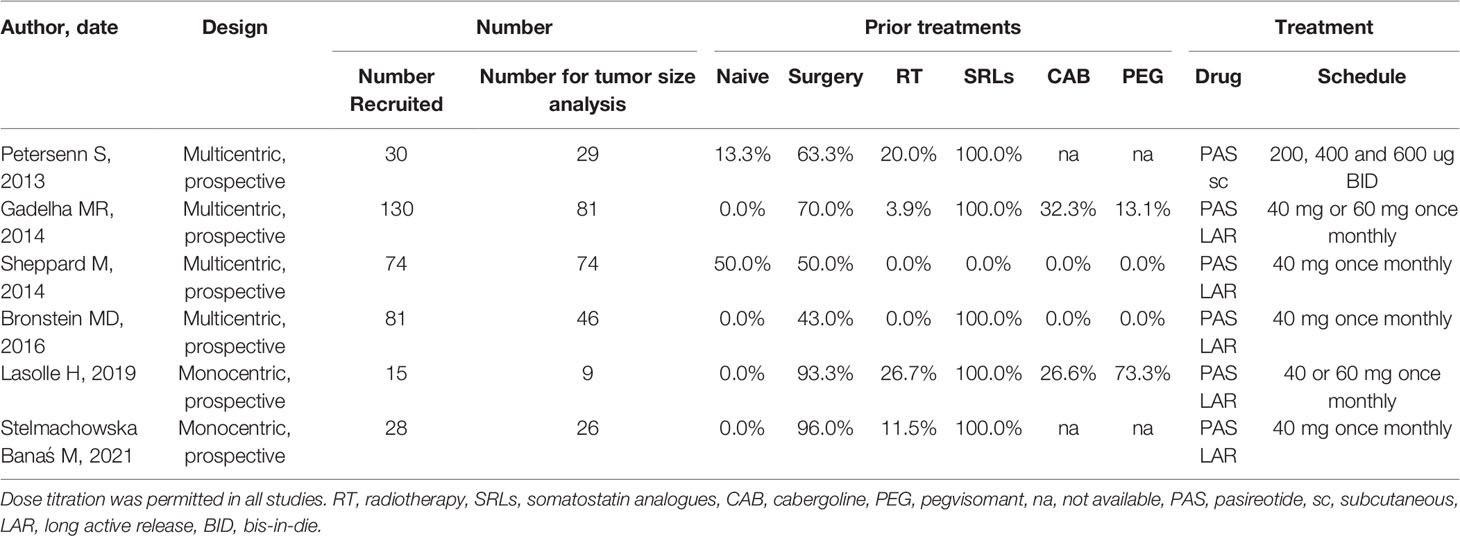 TABLE 1
TABLE 1 Studies considered for the metanalysis in acromegaly.
Table 2
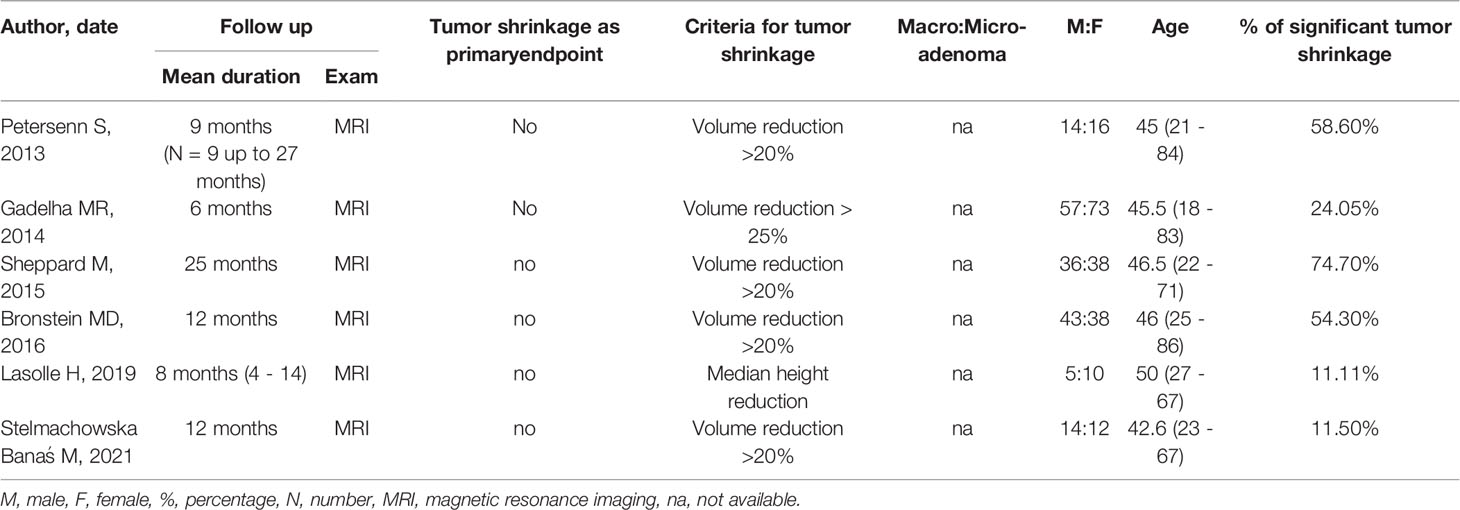 TABLE 2
TABLE 2 Studies considered for the metanalysis in acromegaly.
The study selection process for CD is depicted in Figure 3. The electronic search revealed 122 articles; an additional one had been included post-hoc, through reference analysis of selected articles (N = 123). After the first screening, 91 articles did not meet the eligibility criteria and were discarded. The full-text examination of the remaining studies excluded 29 more articles: 23 did not provide sufficient data on tumor shrinkage, two of them represented the core phase of extension studies, two referred to subsets of patients included in a larger study and two did not provide sufficient data regarding the percentage of tumor size reduction. Thus, three studies fulfilling eligibility criteria (reported in Tables 3, 4) were selected for data extraction and analysis.
Figure 3
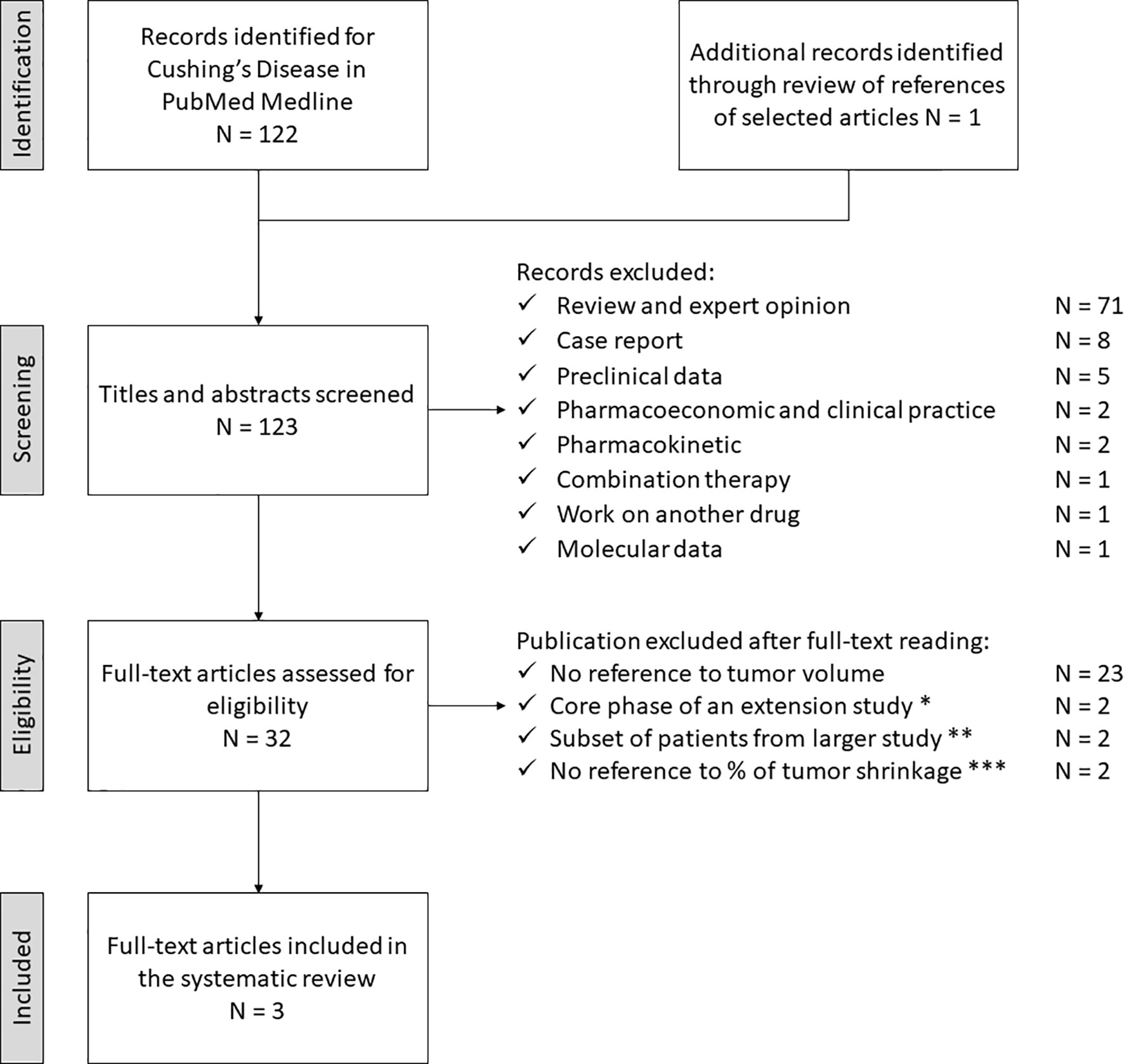 FIGURE 3
FIGURE 3 Search strategy for Cushing’s Disease. * Lacroix A, 2018 (PAS LAR, phase III) and Lacroix A, 2020 (PAS sc, phase III
post-hoc analysis). ** Simeoli C, 2014 (PAS sc) and Colao A 2012 (PAS sc, phase III). *** Daniel E, 2018 (PAS sc and LAR) and Trementino L, 2016 (PAS sc). PAS, pasireotide, sc, subcutaneous, LAR, long acting release.
Table 3
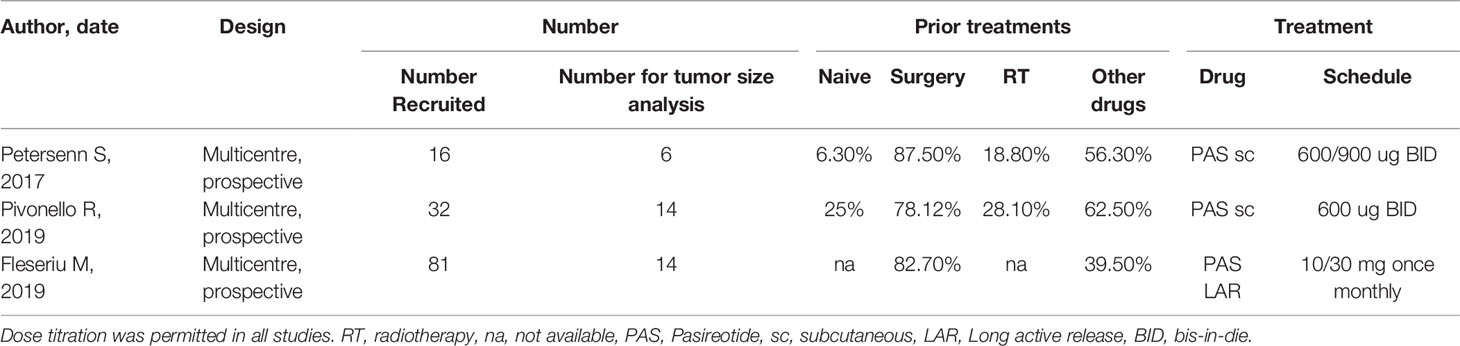 TABLE 3
TABLE 3 Studies considered for the metanalysis in Cushing’s Disease.
Table 4
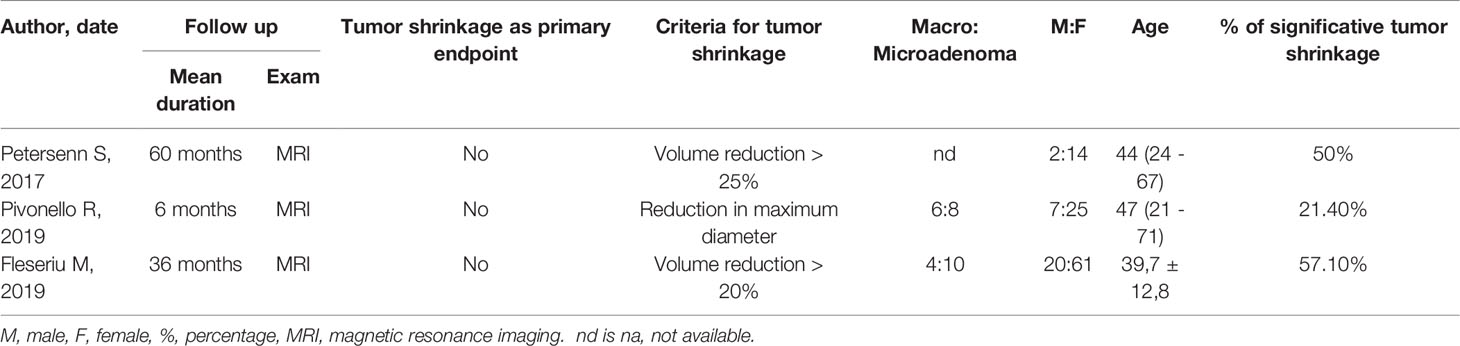 TABLE 4
TABLE 4 Studies considered for the metanalysis in Cushing’s Disease.
Study Characteristics
Four multi- and two mono-centric studies in patients with acromegaly were considered and analyzed, all presenting a prospective design. Tumor size analysis was not one of the primary endpoints in any of the considered studies; from an initial overall recruitment of 358 patients, only 265 were included for tumor size reduction analysis. Most patients had previously undergone different treatments (Table 1). All studies, except one, used PAS LAR, dose titration was allowed in all trials. Median follow-up ranged from 6 to 25 months; MRI was performed to evaluate tumor size reduction and the criteria for considering it significant was mainly based on tumor volume analysis, except for Lasolle H et al. which considered median height reduction (26). Data from the PAOLA study provided separate percentages of significant tumor shrinkage for PAS at 40 mg or 60 mg once monthly; considering that respectively 12 and 7 patients showed a reduction >25%, a significant shrinkage was reported in 19 out of 79 considered cases (24%) (4). Stelmachowska-Banás et al. described one patient with McCune-Albright’s syndrome presenting with pituitary hyperplasia, without a visible adenoma at MRI; as its pituitary volume decreased during treatment, the patient was included in the group with significant tumor shrinkage (27). No study provided information about macro- to micro-adenoma ratio. Data regarding age and male to female ratio are also reported in Table 2.
Three studies including patients with CD met the eligibility criteria (Tables 3, 4); all of them presented a multicentre prospective design, recruiting 139 patients, most of them assuming PAS as a second-line treatment, after a surgical failure. For tumor shrinkage analysis, a subgroup of 34 patients was considered, taking s.c. PAS bis-in-die in two studies and PAS LAR in the third; in all cases titration was admitted. Tumor size analysis was a secondary endpoint in all three studies. Follow-up ranged from 6 to 60 months; tumor size assessment was performed with pituitary MRI. Only Pivonello et al. evaluated maximum diameter, instead of tumor volume changes (28). The population analyzed for tumor shrinkage mainly presented with a microadenoma. Data regarding age and gender are reported in Table 4. In the trial reported by Petersenn S et al., we arbitrarily fixed the criterion to define a significant tumor volume reduction (at least 25% of the baseline size of the pituitary adenoma), and the proportion of responders was calculated from the supplementary materials accordingly (3/6 = 50%) (13). Pivonello et al. separated patients exhibiting mild-moderate from those with severe hypercortisolism; we considered them together for tumor size analysis obtaining an overall proportion of significant size reduction of 21.4% (3 out of 14 subjects) (28).
Risk of Bias
The evaluation of the risk of bias performed with the adapted JBI tool is reported in Table 5. All studies presented clear diagnostic and inclusion criteria, except that of Lasolle H et al. (26). Although all papers reported a valid tool for tumor shrinkage analysis (MRI), two of them did not analyse tumor volume and did not provide a clear definition of significant size reduction (26, 28). Regarding other items, the majority of the considered studies did not appear to present a clear source of bias.
Table 5
 TABLE 5
TABLE 5 Evaluation of the risk of bias performed with the adapted Joanna Briggs’s Institute (JBI) tool.
Meta-Analysis
In the six studies considered for acromegaly, 37.7% (95%CI: [18.7%; 61.5%]) of patients demonstrated a significant tumor size reduction (Figure 4). As expected, heterogeneity in tumor reduction between studies was high (I2 = 90%). We attempted to address publication bias despite the low-number of studies (Figure 6A): Egger’s regression test did not indicate the presence of funnel plot asymmetry (intercept = -3.15 with 95%CI: [-10.17; 3.85], t = -0.883, p = 0.427) and the three-parameter selection model performed for p < 0.05 (and p < 0.1 as a sensitivity analysis) suggested absence of publication bias (28).
Figure 4
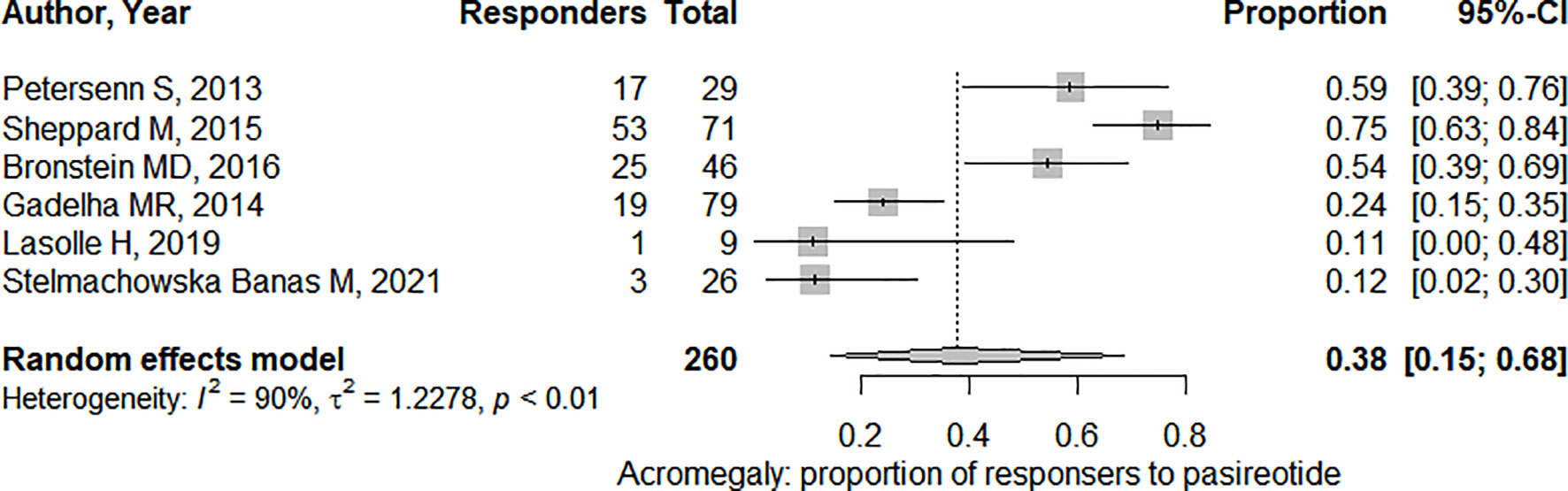 FIGURE 4
FIGURE 4 Pooled effect for the proportion of responders (i.e. presenting significant tumor shrinkage) in acromegaly. CI, confidence interval.
In the three studies considered for CD, 41,2% (95%CI: [22.9%; 62.3%]) of patients overall demonstrated a significant tumor size reduction (Figure 5). The heterogeneity in tumor reduction between the studies represented by I2 amounted to 47%. Publication bias analysis (Figure 6B) was performed using Egger’s regression test (intercept = -1.828 with 95%CI: [-14.53; 10.88], t = -0.282, p = 0.825) without evidence of asymmetry. The three-parameter selection model on the contrary could not be performed due to the small number of studies.
Figure 5
 FIGURE 5
FIGURE 5 Pooled effect for the proportion of responders (i.e. presenting significant tumor shrinkage) in Cushing’s Disease. CI, confidence interval.
Figure 6
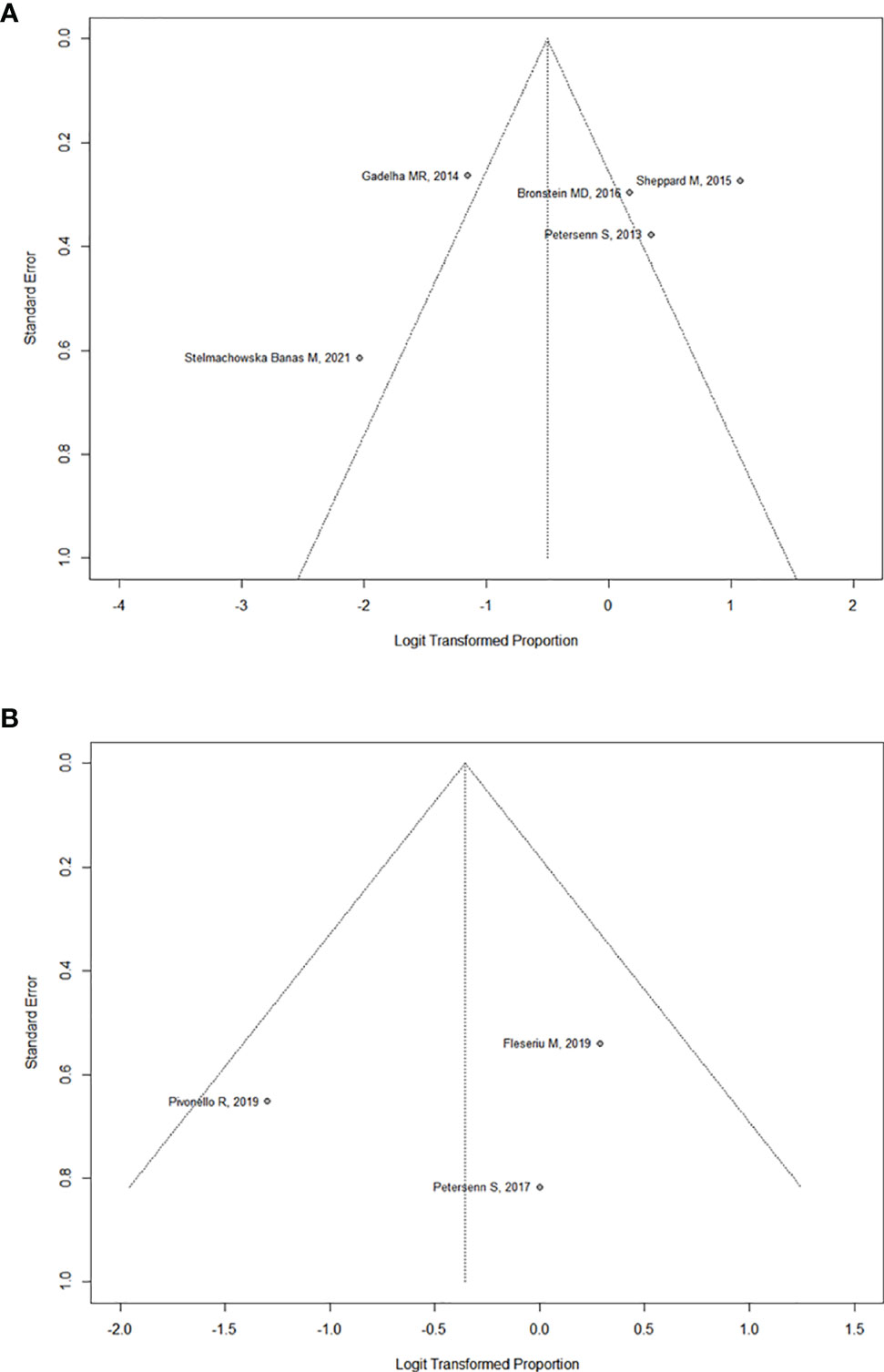 FIGURE 6 (A)
FIGURE 6 (A) Funnel plot assessing publication bias for Acromegaly.
(B) Funnel plot assessing publication bias for Cushing’s Disease.
Discussion
The biochemical efficacy of medical treatment with PAS in GH- or ACTH-secreting pituitary adenomas has been described in previous metanalyses for acromegaly (29, 30) and CD (15), the latter also exploring the clinical benefit. In addition to these reports, this meta-analysis shows that PAS treatment can induce an additional clinically significant tumor shrinkage in approximately 40% of patients.
Acromegaly
Overall, PAS treatment provided tumor shrinkage in 37.7% of the considered patients. A previous metanalysis on octreotide in acromegaly provided a higher percentage of tumor size reduction (over 50%) (20). Nevertheless, since PAS treatment is usually considered as a second- or third-line treatment in the therapeutic flow-chart of acromegaly, the population recruited is mainly composed of patients with first-generation SRL-resistant somatotroph adenomas. This bias in recruited populations of acromegalic patients may explain this difference in the outcome. In a direct comparison, although PAS LAR seemed more effective in achieving biochemical control, both the SRLs, the first- and the second-generation types, achieved similar percentages of tumor shrinkage (6, 7). Moreover, in the crossover extension, the switch from octreotide to PAS was more effective than the reverse schedule, achieving a slightly higher percentage of further significant tumor shrinkage (8). Lasolle et al. reported that the expression of SSR type 5 and the granulation pattern are of limited value for the prediction of PAS responsiveness: 5 out of 9 somatotropinomas in their series were densely granulated (two did not respond to PAS), and the expression of SSR type 5 was modest in one controlled patient (26).
Other than SRLs, a further therapeutic option targeting the somatotroph adenoma is cabergoline, either as monotherapy in mild cases or as an add-on treatment for resistant adenomas (18). In a previous metanalysis, cabergoline in monotherapy resulted less effective than SRLs, achieving tumor shrinkage in about one third of the enrolled patients (31). It should also be mentioned that some studies reported an escape phenomenon from its treatment efficacy (32).
Data coming from the combination of PAS LAR and pegvisomant in acromegaly were not considered in our metanalysis, due to inclusion criteria and variable combination therapy of the two drugs (33). Since some cases of adenoma growth had been reported during pegvisomant use (34, 35), this combination therapy represents a rational approach, but tumor volume analysis is less reliable, given the purpose of our study. Despite concerns regarding tumor growth, pegvisomant effectiveness in acromegaly is well documented (18, 29), although the cost of this combination treatment can limit its applicability in real-life practice. Moreover, it is worth mentioning Coopmans and collaborators’ follow-up analysis, suggesting a PAS mediated anti-tumoral effect in acromegaly. During treatment, patients exhibited a significant increase in T2-weighted sequences signal at MRI; moreover, patients exhibiting this MRI characteristic in their adenomas showed a more evident decrease in IGF-1 levels, but not a similar pattern in reduction of pituitary adenoma size (36). This finding may be related to cell degeneration or tumor cell necrosis, without necessarily determining significant tumor size reduction. Further studies, probably with more data coming from histological reports, may be necessary to better understand these findings.
Cushing’s Disease
Overall, PAS treatment provided significant tumor shrinkage in 41.2% of CD patients. Regarding pituitary-directed drugs, at this moment available for CD treatment, the efficacy of cabergoline has been proven in vitro studies, but its efficacy in clinical trials is still debated (15, 37). In a previous prospective study, cabergoline induced significant tumor shrinkage (defined as tumor volume reduction >20%) in 4 out of 20 (20%) of the patients recruited after 24 months (38). PAS is the only pituitary-directed treatment for this condition approved by Drug Agencies. Although few studies have been considered in this metanalysis, due to the strict inclusion criteria, PAS appears more effective in tumor size reduction versus cabergoline, resulting in a better choice in CD therapy when aiming to control the pituitary adenoma.
In contrast to acromegaly, the majority of CD patients present a microadenoma, suggesting that tumor size might be a less relevant issue during medical treatment, even if the “cure” of the disease may forecast the resolution of the adenoma. Besides, up to 30% of CD patients, depending also on MRI accuracy and neuro-radiologist’s expertise, may present with negative imaging that prevents any evaluation of tumor shrinkage (39). In spite of that, endocrinologists, not so infrequently, deal with aggressive corticotroph adenomas, characterized by invasive local growth and/or resistance to conventional therapies. This challenging entity often requires multidisciplinary expertise to suggest different approaches, including PAS treatment (40). It should be mentioned that some non-pituitary targeting drugs, as inhibitors of cortisol synthesis, have been associated with tumor growth, due to cortisol-ACTH negative feedback. In particular, during osilodrostat treatment in a phase III study, four recruited patients discontinued osilodrostat after a significant increase in tumor volume (two with micro- and two with macro-adenomas 41), and this growth had also been described during ketoconazole and mitotane treatments (42). Thus, it may be speculated that PAS could provide a rational approach as an combination treatment with steroidogenesis inhibitors. Moreover, after bilateral adrenalectomy, pituitary adenoma tumor size is of the utmost importance, as patients may be at risk of developing a progression of the adenoma, the so-called Nelson’s syndrome. In a prospective study from Daniel E et al., PAS proved to be also effective in this setting, reducing ACTH levels and stabilizing the residual tumor over a treatment period of 7 months (43). Further studies, with longer treatment observation, may reveal whether PAS may achieve significant tumor shrinkage in these patients, as suggested by previous case reports in literature (44, 45).
Conclusion
The main limitation of our study resides in the scarce literature provided up to now (260 patients with acromegaly and 34 with CD), in the different therapy schedules and different criteria for tumor shrinkage in the selected study (largest tumor diameter vs a selected percentage of reduction). Moreover, in none of the study tumor reduction was one of the primary endpoints, and surgery was performed before PAS in most patients (78-88% of CD and 43-96% of acromegaly).
PAS is a novel compound, with a rising role in the treatment of secreting pituitary adenomas. Thus, this topic might be amplified with more data coming from further clinical studies, as real-life studies, possibly also addressing markers predictive of response to this treatment (e.g., expression of SSR type 2 and type 5 or somatic mutations in USP8 at tissue level of ACTH-secreting adenomas). Nevertheless, we can already state that PAS treatment is effective in reducing tumor size, especially in acromegaly. Our results strengthen the role of PAS treatment in somatotroph and corticotroph adenomas, especially when tumor volume is a relevant issue (i.e. tumor progression, extrasellar invasion) (18, 39), as a neoadjuvant treatment before surgery or as tailored treatment, alone or in combination, in persistent disease or when surgery is not feasible. Future research aiming to characterize markers predictive of response could help to identify optimal candidates for this treatment.
Data Availability Statement
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding author.
Ethics Statement
Informed consent was obtained from all subjects participating in the studies analyzed.
Author Contributions
Authors involved contributed to research as reported: literature search (FC, AM), preparation of original draft (FC, AM, MB, LD), literature review (CS, FC, AM, MF), manuscript editing (CS, FC, AM, MB, LD, MF) and supervision (RM, CS). All authors approved the final version of the paper.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chinezu L, Vasiljevic A, Jouanneau E, François P, Borda A, Trouillas J, et al. Expression of Somatostatin Receptors, SSTR2A and SSTR5, in 108 Endocrine Pituitary Tumors Using Immunohistochemical Detection With New Specific Monoclonal Antibodies. Hum Pathol (2014) 45(1):71–7. doi: 10.1016/j.humpath.2013.08.007
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Ceccato F, Scaroni C, Boscaro M. Clinical Use of Pasireotide for Cushing’s Disease in Adults. Ther Clin Risk Manage (2015) 11:425–34. doi: 10.2147/TCRM.S37314
CrossRef Full Text | Google Scholar
3. Melmed S, Colao A, Barkan A, Molitch M, Grossman AB, Kleinberg D, et al. Guidelines for Acromegaly Management: An Update. J Clin Endocrinol Metab (2009) 94(5):1509–17. doi: 10.1210/jc.2008-2421
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Gadelha MR, Bronstein MD, Brue T, Coculescu M, Fleseriu M, Guitelman M, et al. Pasireotide Versus Continued Treatment With Octreotide or Lanreotide in Patients With Inadequately Controlled Acromegaly (PAOLA): A Randomised, Phase 3 Trial. Lancet Diabetes Endocrinol (2014) 2(11):875–84. doi: 10.1016/S2213-8587(14)70169-X
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Colao A, Bronstein MD, Brue T, De Marinis L, Fleseriu M, Guitelman M, et al. Pasireotide for Acromegaly: Long-Term Outcomes From an Extension to the Phase III PAOLA Study. Eur J Endocrinol (2020) 182(6):583. doi: 10.1530/EJE-19-0762
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Colao A, Bronstein MD, Freda P, Gu F, Shen CC, Gadelha M, et al. Pasireotide Versus Octreotide in Acromegaly: A Head-to-Head Superiority Study. J Clin Endocrinol Metab (2014) 99(3):791–9. doi: 10.1210/jc.2013-2480
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Sheppard M, Bronstein MD, Freda P, Serri O, De Marinis L, Naves L, et al. Pasireotide LAR Maintains Inhibition of GH and IGF-1 in Patients With Acromegaly for Up to 25 Months: Results From the Blinded Extension Phase of a Randomized, Double-Blind, Multicenter, Phase III Study. [Published Correction Appears in Pituitary]. (2015) 18(3):385–94. doi: 10.1007/s11102-014-0585-6
CrossRef Full Text | Google Scholar
8. Bronstein MD, Fleseriu M, Neggers S, Colao A, Sheppard M, Gu F, et al. Switching Patients With Acromegaly From Octreotide to Pasireotide Improves Biochemical Control: Crossover Extension to a Randomized, Double-Blind, Phase III Study. BMC Endocr Disord (2016) 16:16. doi: 10.1186/s12902-016-0096-8
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Gadelha M, Bex M, Colao A, Pedroza García EM, Poiana C, Jimenez-Sanchez M, et al. Evaluation of the Efficacy and Safety of Switching to Pasireotide in Patients With Acromegaly Inadequately Controlled With First-Generation Somatostatin Analogs. Front Endocrinol (Lausanne). (2020) 10:931. doi: 10.3389/fendo.2019.00931
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Colao A, Petersenn S, Newell-Price J, Findling JW, Gu F, Maldonado M, et al. A 12-Month Phase 3 Study of Pasireotide in Cushing’s Disease. N Engl J Med (2012) 366(10):914–24. doi: 10.1056/NEJMoa1105743
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Pivonello R, Petersenn S, Newell-Price J, Findling JW, Gu F, Maldonado M, et al. Pasireotide Treatment Significantly Improves Clinical Signs and Symptoms in Patients With Cushing’s Disease: Results From a Phase III Study. Clin Endocrinol (Oxf). (2014) 81(3):408–17. doi: 10.1111/cen.12431
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Lacroix A, Gu F, Gallardo W, Pivonello R, Yu Y, Witek P, et al. Efficacy and Safety of Once-Monthly Pasireotide in Cushing’s Disease: A 12 Month Clinical Trial. Lancet Diabetes Endocrinol (2018) 6(1):17–26. doi: 10.1016/S2213-8587(17)30326-1
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Petersenn S, Salgado LR, Schopohl J, Portocarrero-Ortiz L, Arnaldi G, Lacroix A, et al. Long-Term Treatment of Cushing’s Disease With Pasireotide: 5-Year Results From an Open-Label Extension Study of a Phase III Trial. Endocrine. (2017) 57(1):156–65. doi: 10.1007/s12020-017-1316-3
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Fleseriu M, Petersenn S, Biller BMK, Kadioglu P, De Block C, T’Sjoen G, et al. Long-Term Efficacy and Safety of Once-Monthly Pasireotide in Cushing’s Disease: A Phase III Extension Study. Clin Endocrinol (Oxf). (2019) 91(6):776–85. doi: 10.1111/cen.14081
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Simões Corrêa Galendi J, Correa Neto ANS, Demetres M, Boguszewski CL, Nogueira VDSN. Effectiveness of Medical Treatment of Cushing’s Disease: A Systematic Review and Meta-Analysis. Front Endocrinol (Lausanne). (2021) 12:732240. doi: 10.3389/fendo.2021.732240
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Hayashi K, Inoshita N, Kawaguchi K, Ibrahim Ardisasmita A, Suzuki H, Fukuhara N, et al. The USP8 Mutational Status may Predict Drug Susceptibility in Corticotroph Adenomas of Cushing’s Disease. Eur J Endocrinol (2016) 174(2):213–26. doi: 10.1530/EJE-15-0689
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Samson SL, Gu F, Feldt-Rasmussen U, Zhang S, Yu Y, Witek P, et al. Managing Pasireotide-Associated Hyperglycemia: A Randomized, Open-Label, Phase IV Study. Pituitary. (2021) 24(6):887–903. doi: 10.1007/s11102-021-01161-4
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Katznelson L, Laws ER Jr, Melmed S, Molitch ME, Murad MH, Utz A, et al. Acromegaly: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2014) 99(11):3933–51. doi: 10.1210/jc.2014-2700
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Cozzi R, Montini M, Attanasio R, Albizzi M, Lasio G, Lodrini S, et al. Primary Treatment of Acromegaly With Octreotide LAR: A Long-Term (Up to Nine Years) Prospective Study of its Efficacy in the Control of Disease Activity and Tumor Shrinkage. J Clin Endocrinol Metab (2006) 91(4):1397–403. doi: 10.1210/jc.2005-2347
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Giustina A, Mazziotti G, Torri V, Spinello M, Floriani I, Melmed S. Meta-Analysis on the Effects of Octreotide on Tumor Mass in Acromegaly. PloS One (2012) 7(5):e36411. doi: 10.1371/journal.pone.0036411
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Caron PJ, Bevan JS, Petersenn S, Flanagan D, Tabarin A, Prévost G, et al. Tumor Shrinkage With Lanreotide Autogel 120 Mg as Primary Therapy in Acromegaly: Results of a Prospective Multicenter Clinical Trial. J Clin Endocrinol Metab (2014) 99(4):1282–90. doi: 10.1210/jc.2013-3318
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Potorac I, Petrossians P, Daly AF, Alexopoulou O, Borot S, Sahnoun-Fathallah M, et al. T2-Weighted MRI Signal Predicts Hormone and Tumor Responses to Somatostatin Analogs in Acromegaly. Endocr Relat Cancer. (2016) 23(11):871–81. doi: 10.1530/ERC-16-0356
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Schardt C, Adams MB, Owens T, Keitz S, Fontelo P. Utilization of the PICO Framework to Improve Searching PubMed for Clinical Questions. BMC Med Inform Decis Mak. (2007) 7:16. doi: 10.1186/1472-6947-7-16
PubMed Abstract | CrossRef Full Text | Google Scholar
24. McInnes MDF, Moher D, Thombs BD, McGrath TA, Bossuyt PM. Preferred Reporting Items for a Systematic Review and Meta-Analysis of Diagnostic Test Accuracy Studies: The PRISMA-DTA Statement [Published Correction Appears in JAMA. 2019 Nov 26;322(20):2026]. JAMA. (2018) 319(4):388–96. doi: 10.1001/jama.2017.19163
CrossRef Full Text | Google Scholar
25. Munn Z, Barker TH, Moola S, Tufanaru C, Stern C, McArthur A, et al. Methodological Quality of Case Series Studies: An Introduction to the JBI Critical Appraisal Tool. JBI Evid Synth. (2020) 18(10):2127–33. doi: 10.11124/JBISRIR-D-19-00099
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Lasolle H, Ferriere A, Vasiljevic A, Eimer S, Nunes ML, Tabarin A. Pasireotide-LAR in Acromegaly Patients Treated With a Combination Therapy: A Real-Life Study. Endocr Connect. (2019) 8(10):1383–94. doi: 10.1530/EC-19-0332
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Stelmachowska-Banaś M, Czajka-Oraniec I, Tomasik A, Zgliczyński W. Real-World Experience With Pasireotide-LAR in Resistant Acromegaly: A Single Center 1-Year Observation. Pituitary. (2022) 25(1):180–90. doi: 10.1007/s11102-021-01185-w
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Pivonello R, Arnaldi G, Scaroni C, Giordano C, Cannavò S, Iacuaniello D, et al. The Medical Treatment With Pasireotide in Cushing’s Disease: An Italian Multicentre Experience Based on “Real-World Evidence”. Endocrine. (2019) 64(3):657–72. doi: 10.1007/s12020-018-1818-7
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Leonart LP, Ferreira VL, Tonin FS, Fernandez-Llimos F, Pontarolo R. Medical Treatments for Acromegaly: A Systematic Review and Network Meta-Analysis. Value Health (2018) 21(7):874–80. doi: 10.1016/j.jval.2017.12.014”
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Qiao N, He M, Shen M, Zhang Q, Zhang Z, Shou X, et al. Comparative Efficacy Of Medical Treatment For Acromegaly: A Systematic Review And Network Meta-Analysis Of Integrated Randomized Trials And Observational Studies. Endocr Pract (2020) 26(4):454–62. doi: 10.4158/EP-2019-0528
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Muhammad A, van der Lely AJ, Delhanty PJD, Dallenga AHG, Haitsma IK, Janssen JAMJL, et al. Efficacy and Safety of Switching to Pasireotide in Patients With Acromegaly Controlled With Pegvisomant and First-Generation Somatostatin Analogues (PAPE Study). J Clin Endocrinol Metab (2018) 103(2):586–95. doi: 10.1210/jc.2017-02017
PubMed Abstract | CrossRef Full Text | Google Scholar
34. uhk JH, Jung S, Psychogios MN, Göricke S, Hartz S, Schulz-Heise S, et al. Tumor Volume of Growth Hormone-Secreting Pituitary Adenomas During Treatment With Pegvisomant: A Prospective Multicenter Study. J Clin Endocrinol Metab (2010) 95(2):552–8. doi: 10.1210/jc.2009-1239
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Marazuela M, Paniagua AE, Gahete MD, Lucas T, Alvarez-Escolá C, Manzanares R, et al. Somatotroph Tumor Progression During Pegvisomant Therapy: A Clinical and Molecular Study. J Clin Endocrinol Metab (2011) 96(2):E251–9. doi: 10.1210/jc.2010-1742
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Coopmans EC, van der Lely AJ, Schneiders JJ, Neggers SJCMM. Potential Antitumour Activity of Pasireotide on Pituitary Tumours in Acromegaly. Lancet Diabetes Endocrinol (2019) 7(6):425–6. doi: 10.1016/S2213-8587(19)30113-5
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Ferriere A, Cortet C, Chanson P, Delemer B, Caron P, Chabre O, et al. Cabergoline for Cushing’s Disease: A Large Retrospective Multicenter Study. Eur J Endocrinol (2017) 176(3):305–14. doi: 10.1530/EJE-16-0662
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Pivonello R, De Martino MC, Cappabianca P, De Leo M, Faggiano A, Lombardi G, et al. The Medical Treatment of Cushing’s Disease: Effectiveness of Chronic Treatment With the Dopamine Agonist Cabergoline in Patients Unsuccessfully Treated by Surgery. J Clin Endocrinol Metab (2009) 94(1):223–30. doi: 10.1210/jc.2008-1533
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Fleseriu M, Auchus R, Bancos I, Ben-Shlomo A, Bertherat J, Biermasz NR, et al. Consensus on Diagnosis and Management of Cushing’s Disease: A Guideline Update. Lancet Diabetes Endocrinol (2021) 9(12):847–75. doi: 10.1016/S2213-8587(21)00235-7
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Ceccato F, Lombardi G, Manara R, Emanuelli E, Denaro L, Milanese L, et al. Temozolomide and Pasireotide Treatment for Aggressive Pituitary Adenoma: Expertise at a Tertiary Care Center. J Neurooncol. (2015) 122(1):189–96. doi: 10.1007/s11060-014-1702-0
PubMed Abstract | CrossRef Full Text | Google Scholar
41. Pivonello R, Fleseriu M, Newell-Price J, Bertagna X, Findling J, Shimatsu A, et al. Efficacy and Safety of Osilodrostat in Patients With Cushing’s Disease (LINC 3): A Multicentre Phase III Study With a Double-Blind, Randomised Withdrawal Phase. Lancet Diabetes Endocrinol (2020) 8(9):748–61. doi: 10.1016/S2213-8587(20)30240-0
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Varlamov EV, Han AJ, Fleseriu M. Updates in Adrenal Steroidogenesis Inhibitors for Cushing’s Syndrome – A Practical Guide. Best Pract Res Clin Endocrinol Metab (2021) 35(1):101490. doi: 10.1016/j.beem.2021.101490
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Daniel E, Debono M, Caunt S, Girio-Fragkoulakis C, Walters SJ, Akker SA, et al. A Prospective Longitudinal Study of Pasireotide in Nelson’s Syndrome. Pituitary. (2018) 21(3):247–55. doi: 10.1007/s11102-017-0853-3
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Katznelson L. Sustained Improvements in Plasma ACTH and Clinical Status in a Patient With Nelson’s Syndrome Treated With Pasireotide LAR, a Multireceptor Somatostatin Analog. J Clin Endocrinol Metab (2013) 98(5):1803–7. doi: 10.1210/jc.2013-1497
PubMed Abstract | CrossRef Full Text | Google Scholar
45. He X, Spencer-Segal JL. Rapid Response of Nelson’s Syndrome to Pasireotide in Radiotherapy-Naive Patient. Clin Diabetes Endocrinol (2020) 6(1):22. doi: 10.1186/s40842-020-00110-7
PubMed Abstract | CrossRef Full Text | Google Scholar



